Department of Genetics and Center for Epigenomics, Albert Einstein College of Medicine, Bronx, New York, United States of America.
Department of Pediatrics, Albert Einstein College of Medicine, Bronx, New York, United States of America.
PLoS One. 2019 Apr 25;14(4):e0215987. doi: 10.1371/journal.pone.0215987. eCollection 2019.
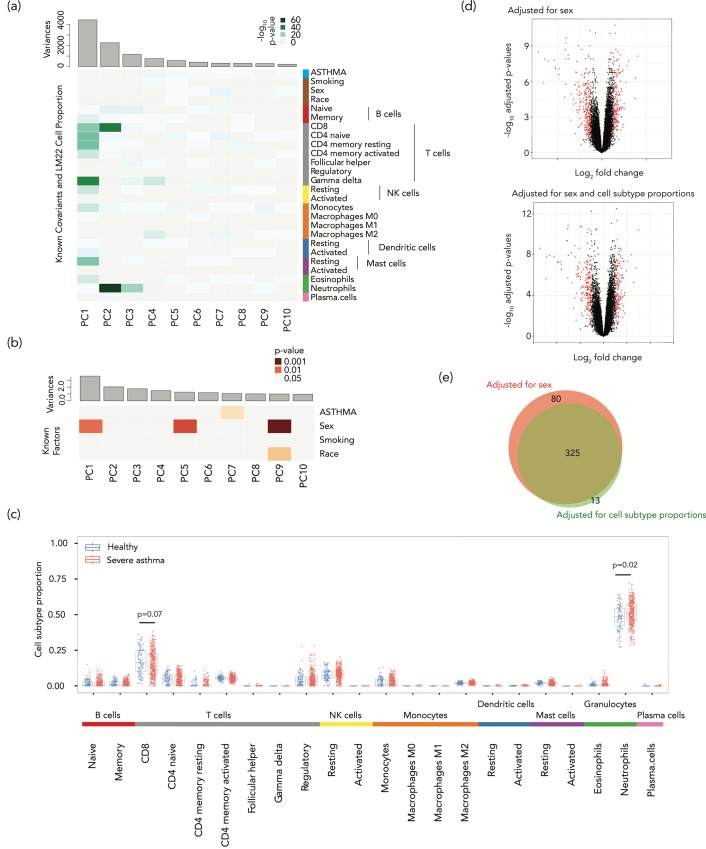
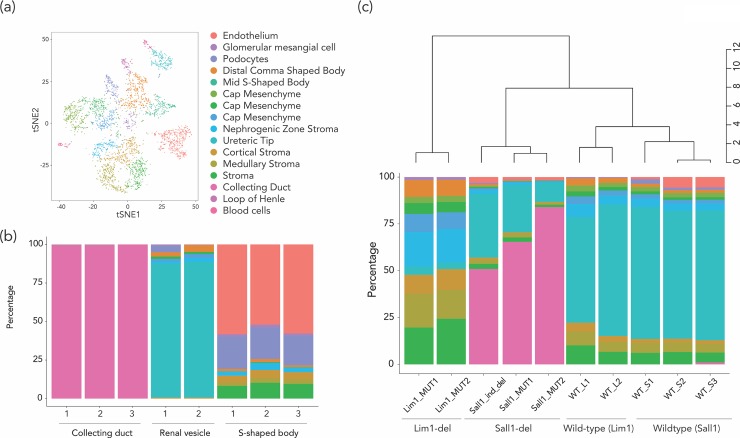
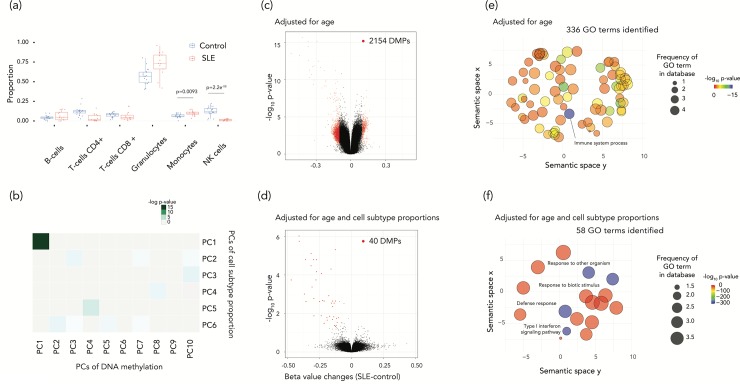
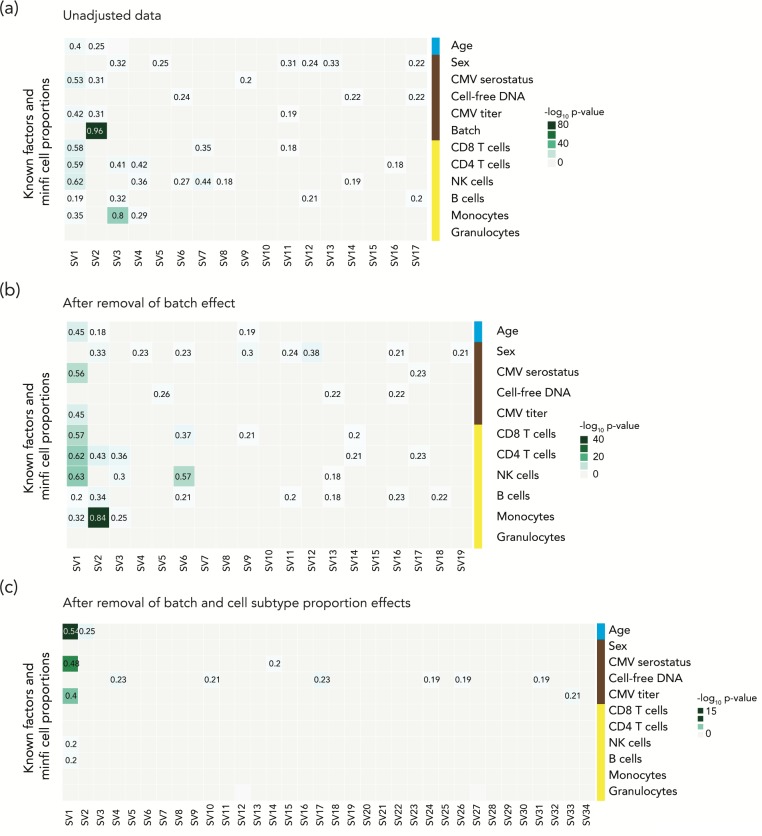
Cell subtype proportion variability between samples contributes significantly to the variation of functional genomic properties such as gene expression or DNA methylation. Although the impact of the variation of cell subtype composition on measured genomic quantities is recognized, and some innovative tools have been developed for the analysis of heterogeneous samples, most functional genomics studies using samples with mixed cell types still ignore the influence of cell subtype proportion variation, or just deal with it as a nuisance variable to be eliminated. Here we demonstrate how harvesting information about cell subtype proportions from functional genomics data can provide insights into cellular changes associated with phenotypes. We focused on two types of mixed cell populations, human blood and mouse kidney. Cell type prediction is well developed in the former, but not currently in the latter. Estimating the cellular repertoire is easier when a reference dataset from purified samples of all cell types in the tissue is available, as is the case for blood. However, reference datasets are not available for most other tissues, such as the kidney. In this study, we showed that the proportion of alterations attributable to changes in the cellular composition varies strikingly in the two disorders (asthma and systemic lupus erythematosus), suggesting that the contribution of cell subtype proportion changes to functional genomic properties can be disease-specific. We also showed that a reference dataset from a single-cell RNA-seq study successfully estimated the cell subtype proportions in mouse kidney and allowed us to distinguish altered cell subtype differences between two different knock-out mouse models, both of which had reported a reduced number of glomeruli compared to their wild-type counterparts. These findings demonstrate that testing for changes in cell subtype proportions between conditions can yield important insights in functional genomics studies.
样本间细胞亚型比例的变化对功能基因组特性(如基因表达或 DNA 甲基化)的变化有重要影响。尽管已经认识到细胞亚型组成变化对测量基因组数量的影响,并且已经开发了一些用于分析异质样本的创新工具,但大多数使用混合细胞类型样本的功能基因组学研究仍然忽略了细胞亚型比例变化的影响,或者只是将其视为需要消除的干扰变量。在这里,我们展示了如何从功能基因组学数据中获取有关细胞亚型比例的信息,从而深入了解与表型相关的细胞变化。我们专注于两种混合细胞群体,即人类血液和小鼠肾脏。前者的细胞类型预测已经很成熟,但后者目前还没有。当组织中所有细胞类型的纯化样本的参考数据集可用时,估计细胞组成更容易,如血液。然而,对于大多数其他组织,如肾脏,并没有参考数据集。在这项研究中,我们表明,归因于细胞组成变化的改变比例在两种疾病(哮喘和系统性红斑狼疮)中差异显著,这表明细胞亚型比例变化对功能基因组特性的贡献可能是疾病特异性的。我们还表明,来自单细胞 RNA-seq 研究的参考数据集成功估计了小鼠肾脏的细胞亚型比例,并允许我们区分两种不同敲除小鼠模型之间改变的细胞亚型差异,这两种模型与野生型相比,肾小球数量都减少了。这些发现表明,检测条件之间细胞亚型比例的变化可以在功能基因组学研究中产生重要的见解。