Department of Clinical Pharmacology, School of Pharmacy, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel.
Cloud Pharmaceuticals Inc., Durham, NC 27709, USA.
Int J Mol Sci. 2019 May 5;20(9):2210. doi: 10.3390/ijms20092210.
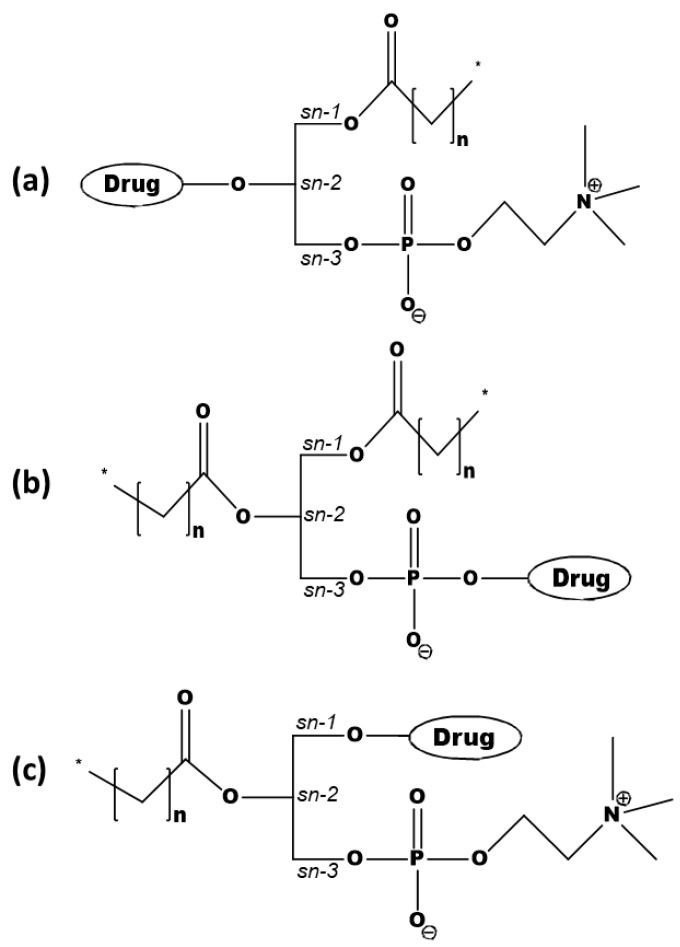
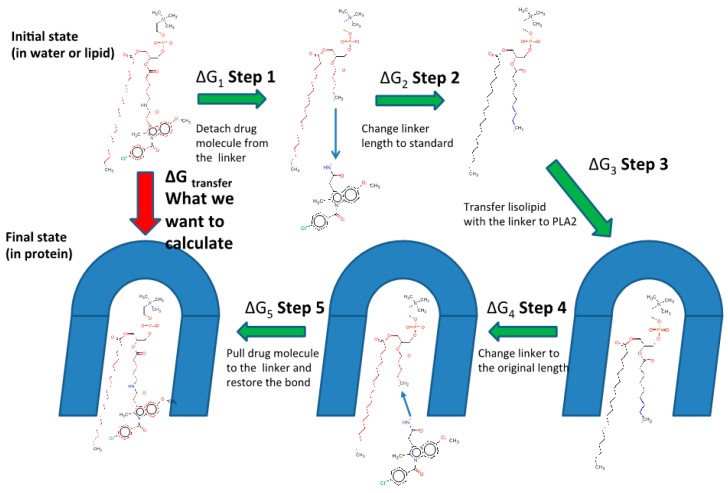
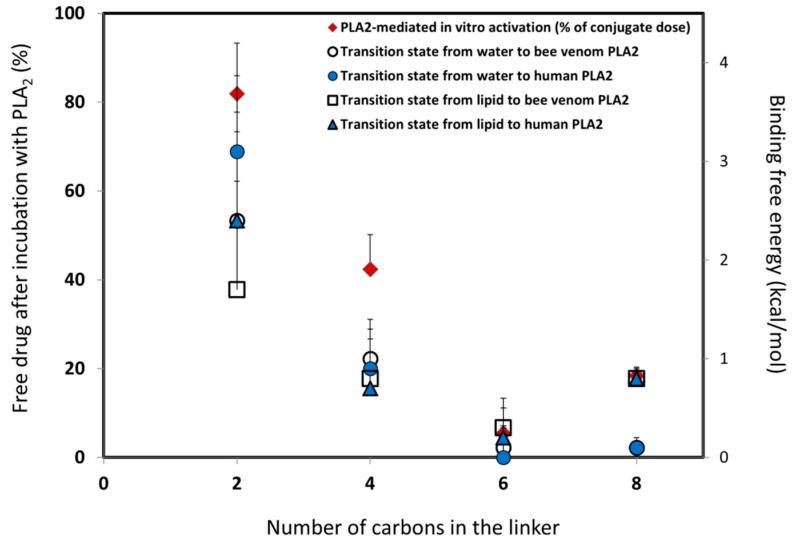
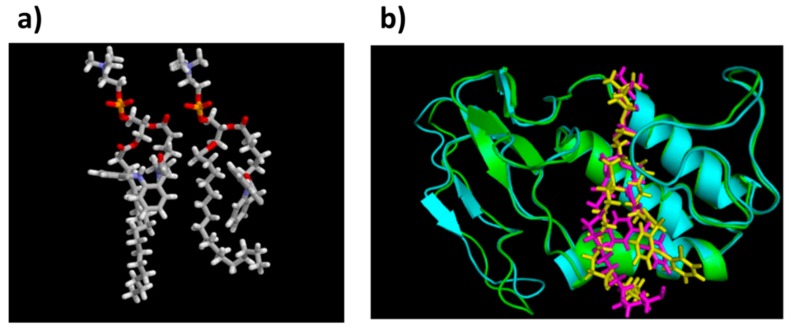
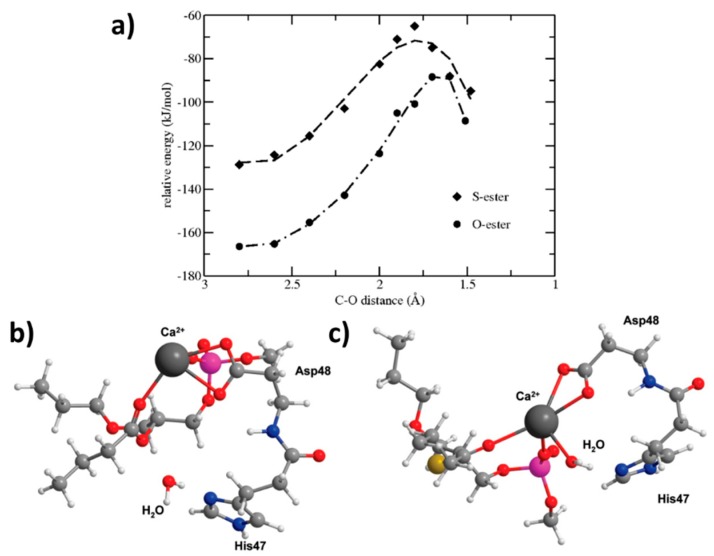
The lipidic prodrug approach is an emerging field for improving a number of biopharmaceutical and drug delivery aspects. Owing to their structure and nature, phospholipid (PL)-based prodrugs may join endogenous lipid processing pathways, and hence significantly improve the pharmacokinetics and/or bioavailability of the drug. Additional advantages of this approach include drug targeting by enzyme-triggered drug release, blood-brain barrier permeability, lymphatic targeting, overcoming drug resistance, or enabling appropriate formulation. The PL-prodrug design includes various structural modalities-different conjugation strategies and/or the use of linkers between the PL and the drug moiety, which considerably influence the prodrug characteristics and the consequent effects. In this article, we describe how molecular modeling can guide the structural design of PL-based prodrugs. Computational simulations can predict the extent of phospholipase A (PLA)-mediated activation, and facilitate prodrug development. Several computational methods have been used to facilitate the design of the pro-drugs, which will be reviewed here, including molecular docking, the free energy perturbation method, molecular dynamics simulations, and free density functional theory. Altogether, the studies described in this article indicate that computational simulation-guided PL-based prodrug molecular design correlates well with the experimental results, allowing for more mechanistic and less empirical development. In the future, the use of molecular modeling techniques to predict the activity of PL-prodrugs should be used earlier in the development process.
脂质前药方法是改善许多生物制药和药物传递方面的新兴领域。由于其结构和性质,基于磷脂 (PL) 的前药可能会加入内源性脂质处理途径,从而显著改善药物的药代动力学和/或生物利用度。这种方法的额外优点包括通过酶触发药物释放、血脑屏障通透性、淋巴靶向、克服药物耐药性或实现适当配方进行药物靶向。PL-前药设计包括各种结构模式-不同的缀合策略和/或在 PL 和药物部分之间使用连接子,这会极大地影响前药的特性和随后的效果。在本文中,我们描述了分子建模如何指导基于 PL 的前药的结构设计。计算模拟可以预测磷脂酶 A (PLA)介导的激活程度,并促进前药的开发。已经使用了几种计算方法来促进前药的设计,本文将对此进行综述,包括分子对接、自由能微扰法、分子动力学模拟和自由密度泛函理论。总的来说,本文中描述的研究表明,基于计算模拟的 PL 前药分子设计与实验结果相关性良好,允许进行更具机制性和更少经验性的开发。在未来,应在开发过程的早期更早地使用分子建模技术来预测 PL-前药的活性。