Department of Pediatric Kidney, Liver and Metabolic Diseases, Hannover Medical School, Hannover, Germany.
Center for Congenital Kidney Diseases, Center for Rare Diseases, Hannover Medical School, Hannover, Germany.
Nat Rev Nephrol. 2019 Jul;15(7):435-455. doi: 10.1038/s41581-019-0152-5.
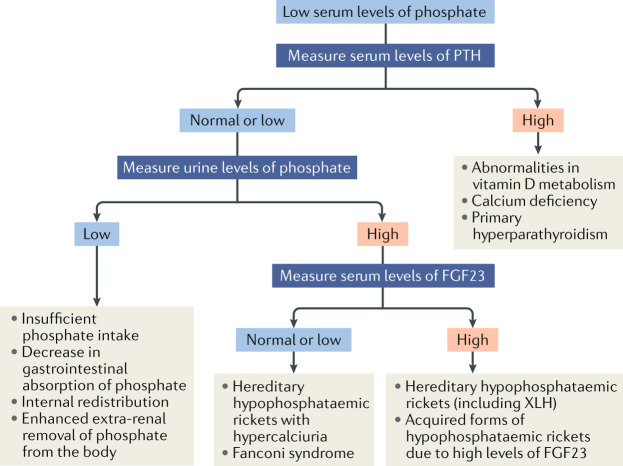
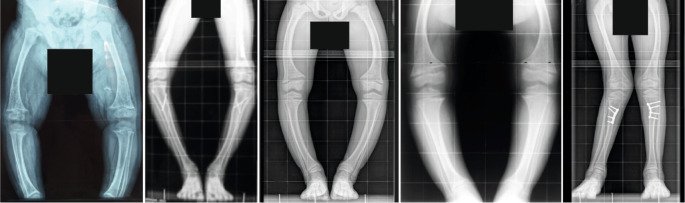
X-linked hypophosphataemia (XLH) is the most common cause of inherited phosphate wasting and is associated with severe complications such as rickets, lower limb deformities, pain, poor mineralization of the teeth and disproportionate short stature in children as well as hyperparathyroidism, osteomalacia, enthesopathies, osteoarthritis and pseudofractures in adults. The characteristics and severity of XLH vary between patients. Because of its rarity, the diagnosis and specific treatment of XLH are frequently delayed, which has a detrimental effect on patient outcomes. In this Evidence-Based Guideline, we recommend that the diagnosis of XLH is based on signs of rickets and/or osteomalacia in association with hypophosphataemia and renal phosphate wasting in the absence of vitamin D or calcium deficiency. Whenever possible, the diagnosis should be confirmed by molecular genetic analysis or measurement of levels of fibroblast growth factor 23 (FGF23) before treatment. Owing to the multisystemic nature of the disease, patients should be seen regularly by multidisciplinary teams organized by a metabolic bone disease expert. In this article, we summarize the current evidence and provide recommendations on features of the disease, including new treatment modalities, to improve knowledge and provide guidance for diagnosis and multidisciplinary care.
X 连锁低磷血症(XLH)是遗传性磷酸盐丢失的最常见原因,与严重并发症相关,如佝偻病、下肢畸形、疼痛、牙齿矿化不良以及儿童不成比例的身材矮小,以及成人生长激素过多、骨软化症、腱病、骨关节炎和假性骨折。XLH 的特征和严重程度在患者之间存在差异。由于其罕见性,XLH 的诊断和特定治疗经常被延迟,这对患者的结果产生不利影响。在本循证指南中,我们建议 XLH 的诊断基于佝偻病和/或骨软化症的迹象,以及在没有维生素 D 或钙缺乏的情况下存在低磷血症和肾脏磷酸盐丢失。只要有可能,在治疗前应通过分子遗传学分析或成纤维细胞生长因子 23(FGF23)水平的测量来确认诊断。由于疾病的多系统性质,患者应由代谢性骨病专家组织的多学科团队定期进行检查。在本文中,我们总结了目前的证据,并就疾病的特征提供了建议,包括新的治疗方式,以提高知识水平,并为诊断和多学科护理提供指导。