School of Medicine, National University of Ireland, Galway, University Road, Galway, Ireland.
Luxembourg Centre for Systems Biomedicine, University of Luxembourg, Belvaux, Luxembourg.
Microbiome. 2019 May 15;7(1):75. doi: 10.1186/s40168-019-0689-3.
The human gut microbiome performs important functions in human health and disease. A classic example for host-gut microbial co-metabolism is host biosynthesis of primary bile acids and their subsequent deconjugation and transformation by the gut microbiome. To understand these system-level host-microbe interactions, a mechanistic, multi-scale computational systems biology approach that integrates the different types of omic data is needed. Here, we use a systematic workflow to computationally model bile acid metabolism in gut microbes and microbial communities.
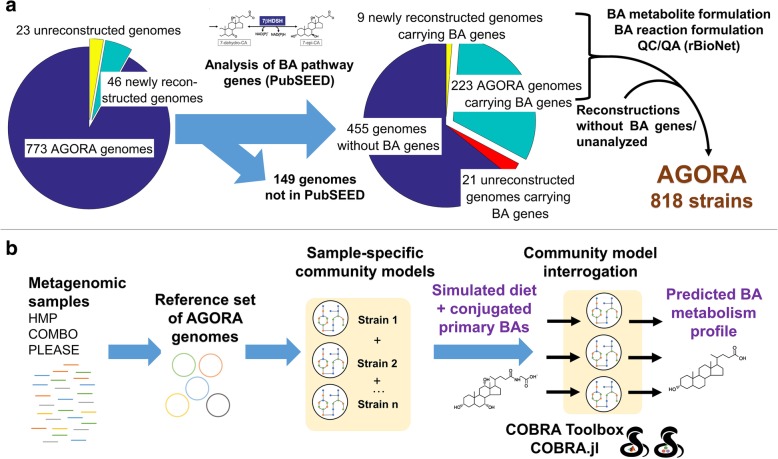
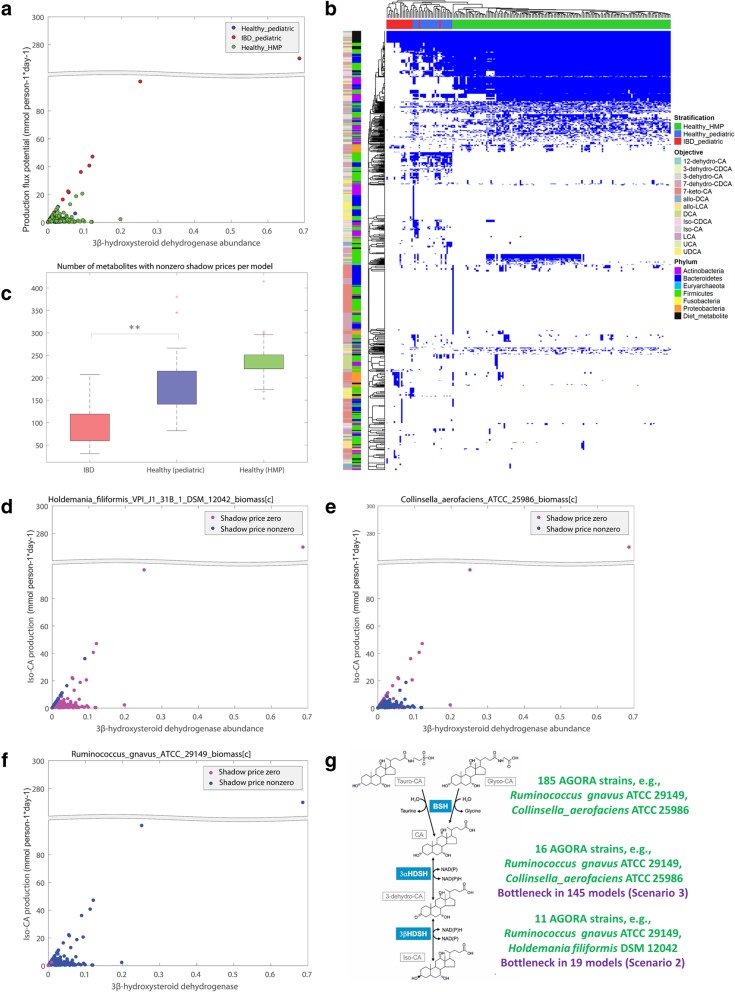
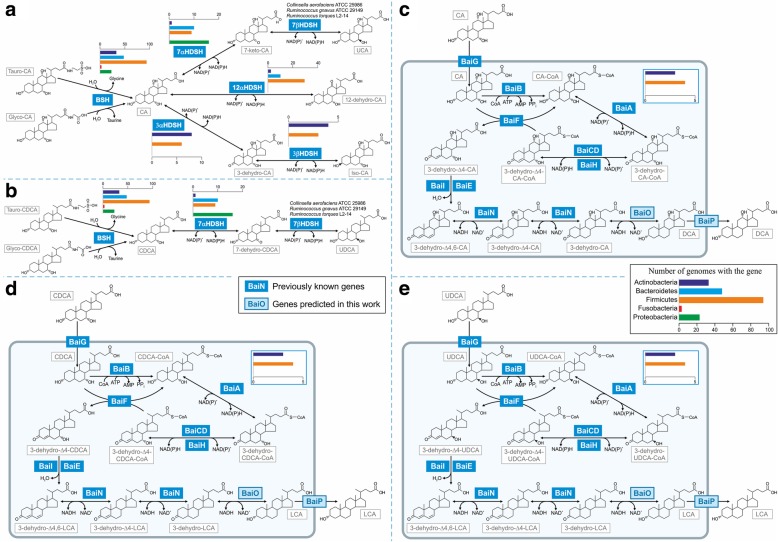
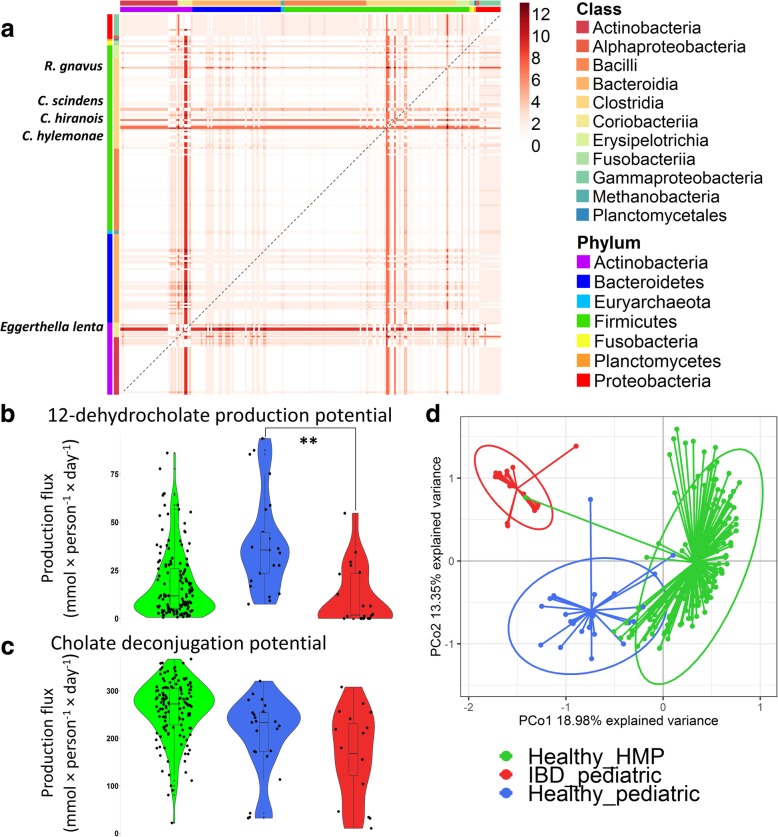
Therefore, we first performed a comparative genomic analysis of bile acid deconjugation and biotransformation pathways in 693 human gut microbial genomes and expanded 232 curated genome-scale microbial metabolic reconstructions with the corresponding reactions (available at https://vmh.life ). We then predicted the bile acid biotransformation potential of each microbe and in combination with other microbes. We found that each microbe could produce maximally six of the 13 secondary bile acids in silico, while microbial pairs could produce up to 12 bile acids, suggesting bile acid biotransformation being a microbial community task. To investigate the metabolic potential of a given microbiome, publicly available metagenomics data from healthy Western individuals, as well as inflammatory bowel disease patients and healthy controls, were mapped onto the genomes of the reconstructed strains. We constructed for each individual a large-scale personalized microbial community model that takes into account strain-level abundances. Using flux balance analysis, we found considerable variation in the potential to deconjugate and transform primary bile acids between the gut microbiomes of healthy individuals. Moreover, the microbiomes of pediatric inflammatory bowel disease patients were significantly depleted in their bile acid production potential compared with that of controls. The contributions of each strain to overall bile acid production potential across individuals were found to be distinct between inflammatory bowel disease patients and controls. Finally, bottlenecks limiting secondary bile acid production potential were identified in each microbiome model.
This large-scale modeling approach provides a novel way of analyzing metagenomics data to accelerate our understanding of the metabolic interactions between the host and gut microbiomes in health and diseases states. Our models and tools are freely available to the scientific community.
人类肠道微生物组在人类健康和疾病中发挥着重要作用。宿主-肠道微生物共代谢的一个经典例子是宿主生物合成初级胆汁酸,然后由肠道微生物组对其进行去结合和转化。为了理解这些系统水平的宿主-微生物相互作用,需要一种整合不同类型组学数据的机制、多尺度计算系统生物学方法。在这里,我们使用一种系统的工作流程来计算模拟肠道微生物和微生物群落中的胆汁酸代谢。
因此,我们首先对 693 个人类肠道微生物基因组中的胆汁酸去结合和生物转化途径进行了比较基因组分析,并在 232 个经过验证的基因组规模微生物代谢重建中扩展了相应的反应(可在 https://vmh.life 上获得)。然后,我们预测了每个微生物的胆汁酸生物转化潜力,并结合其他微生物进行了预测。我们发现,每个微生物在计算机上最多可以产生 13 种次级胆汁酸中的 6 种,而微生物对可以产生多达 12 种胆汁酸,这表明胆汁酸生物转化是微生物群落的一项任务。为了研究给定微生物组的代谢潜力,我们将来自健康西方个体、炎症性肠病患者和健康对照的公开可用的宏基因组数据映射到重建菌株的基因组上。我们为每个人构建了一个大规模的个性化微生物群落模型,该模型考虑了菌株水平的丰度。通过通量平衡分析,我们发现健康个体的肠道微生物组在去结合和转化初级胆汁酸的潜力方面存在相当大的差异。此外,与对照组相比,儿科炎症性肠病患者的微生物组在胆汁酸产生潜力方面明显减少。在个体之间,每个菌株对整体胆汁酸产生潜力的贡献在炎症性肠病患者和对照组之间是不同的。最后,在每个微生物群落模型中都确定了限制次级胆汁酸产生潜力的瓶颈。
这种大规模建模方法为分析宏基因组数据提供了一种新方法,有助于加速我们对健康和疾病状态下宿主和肠道微生物组之间代谢相互作用的理解。我们的模型和工具可供科学界免费使用。