Division of Cell Signalling and Immunology, Sir James Black Centre, School of Life Sciences, University of Dundee, Dundee, DD1 5EH, UK.
MRC Protein Phosphorylation and Ubiquitylation Unit, Sir James Black Centre, School of Life Sciences, University of Dundee, Dundee, DD1 5EH, UK.
Cell Mol Life Sci. 2019 Dec;76(24):4887-4904. doi: 10.1007/s00018-019-03148-8. Epub 2019 May 17.
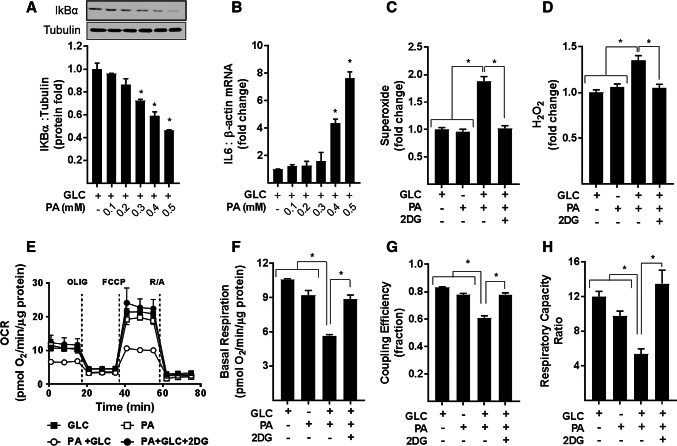
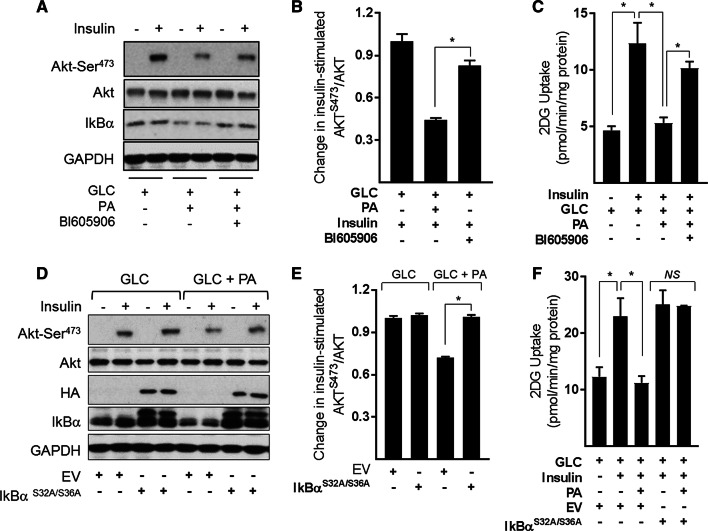
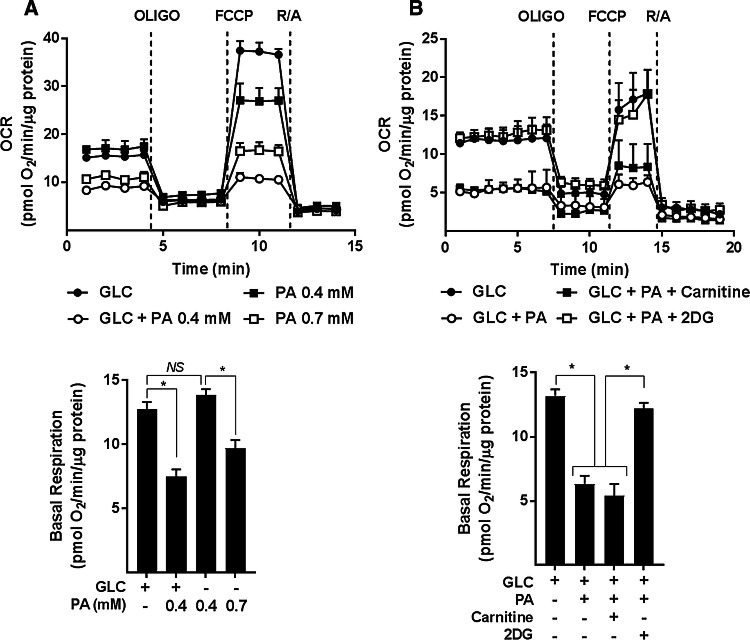
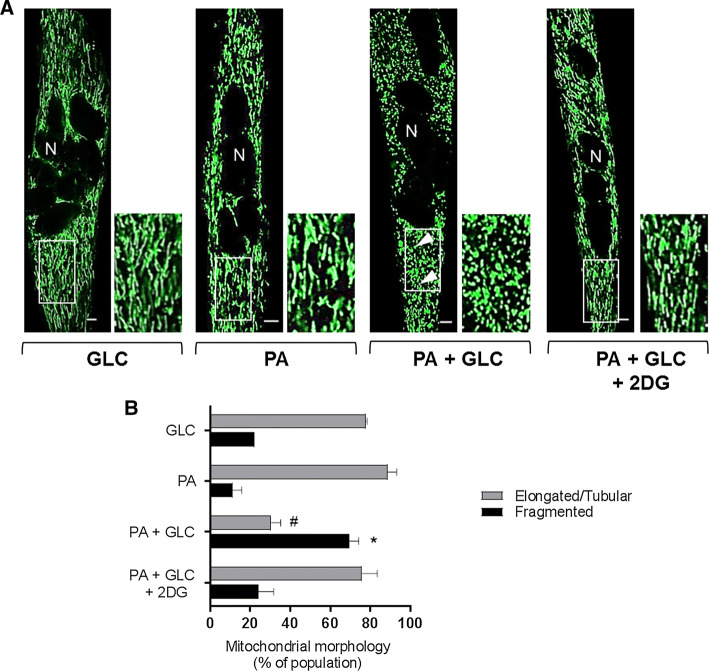
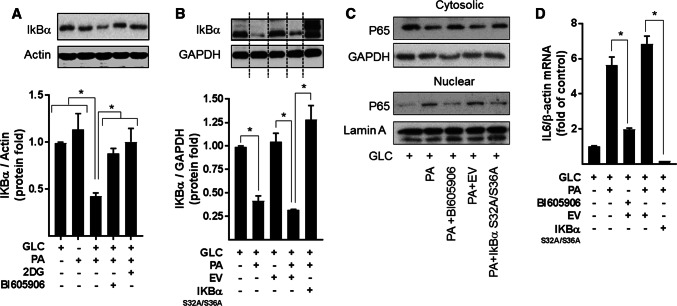
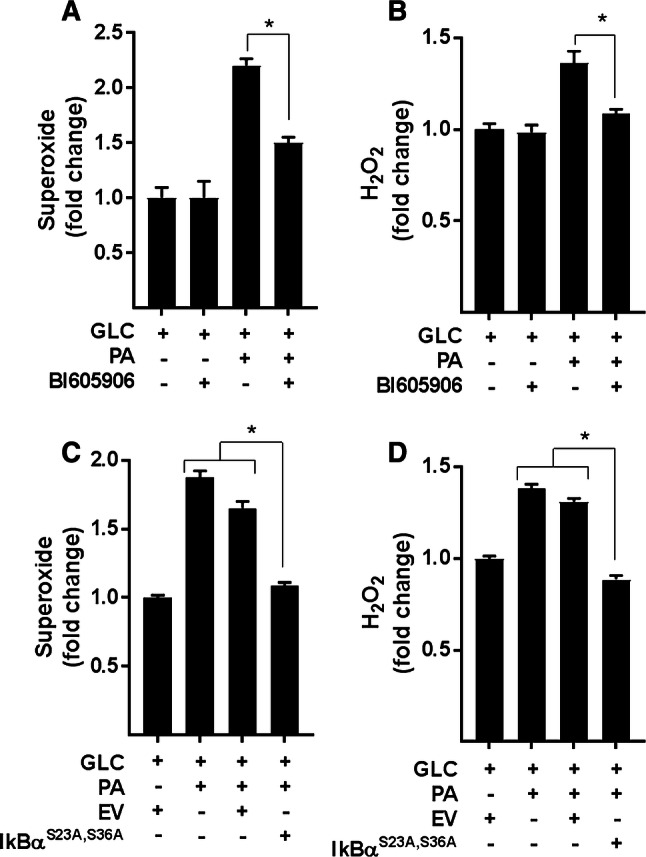
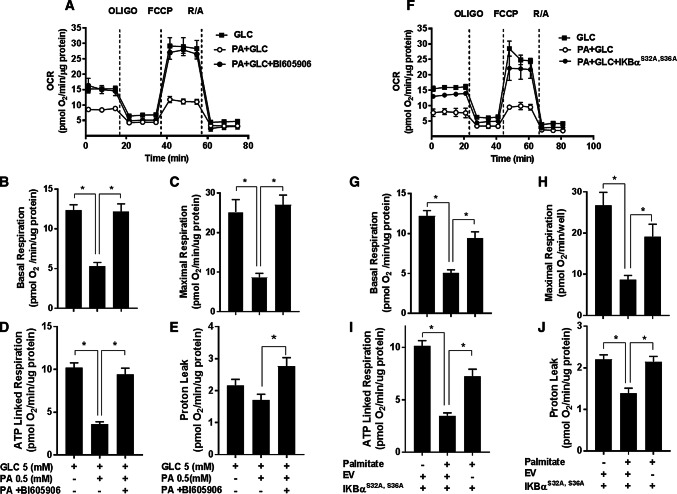
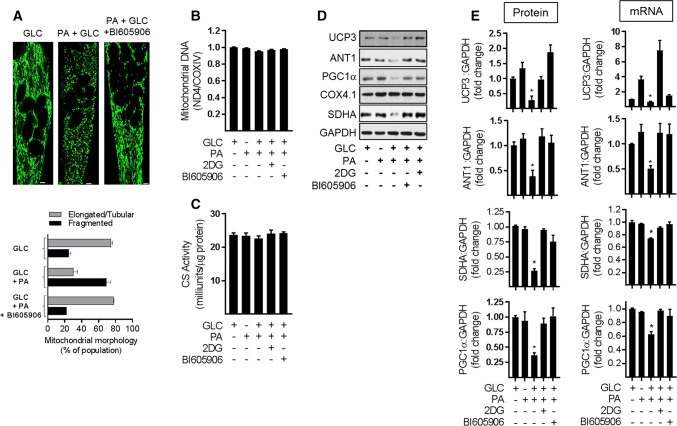
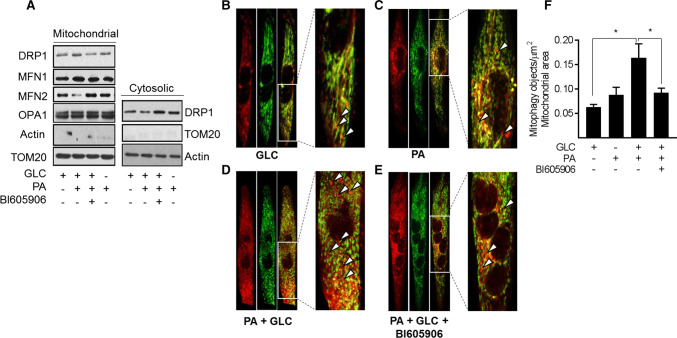
Sustained nutrient (fuel) excess, as occurs during obesity and diabetes, has been linked to increased inflammation, impaired mitochondrial homeostasis, lipotoxicity, and insulin resistance in skeletal muscle. Precisely how mitochondrial dysfunction is initiated and whether it contributes to insulin resistance in this tissue remains a poorly resolved issue. Herein, we examine the contribution that an increase in proinflammatory NFkB signalling makes towards regulation of mitochondrial bioenergetics, morphology, and dynamics and its impact upon insulin action in skeletal muscle cells subject to chronic fuel (glucose and palmitate) overloading. We show sustained nutrient excess of L6 myotubes promotes activation of the IKKβ-NFkB pathway (as judged by a six-fold increase in IL-6 mRNA expression; an NFkB target gene) and that this was associated with a marked reduction in mitochondrial respiratory capacity (>50%), a three-fold increase in mitochondrial fragmentation and 2.5-fold increase in mitophagy. Under these circumstances, we also noted a reduction in the mRNA and protein abundance of PGC1α and that of key mitochondrial components (SDHA, ANT-1, UCP3, and MFN2) as well as an increase in cellular ROS and impaired insulin action in myotubes. Strikingly, pharmacological or genetic repression of NFkB activity ameliorated disturbances in mitochondrial respiratory function/morphology, attenuated loss of SDHA, ANT-1, UCP3, and MFN2 and mitigated the increase in ROS and the associated reduction in myotube insulin sensitivity. Our findings indicate that sustained oversupply of metabolic fuel to skeletal muscle cells induces heightened NFkB signalling and that this serves as a critical driver for disturbances in mitochondrial function and morphology, redox status, and insulin signalling.
持续的营养(燃料)过剩,如肥胖和糖尿病时发生的情况,与骨骼肌中的炎症增加、线粒体动态平衡受损、脂毒性和胰岛素抵抗有关。线粒体功能障碍是如何启动的,以及它是否导致组织中的胰岛素抵抗,这仍然是一个悬而未决的问题。在此,我们研究了促炎 NFkB 信号的增加对线粒体生物能学、形态和动力学的调节作用,以及它对慢性燃料(葡萄糖和棕榈酸)超负荷的骨骼肌细胞中胰岛素作用的影响。我们发现,L6 肌管的持续营养过剩会促进 IKKβ-NFkB 途径的激活(通过 IL-6 mRNA 表达增加六倍来判断;NFkB 靶基因),这与线粒体呼吸能力的显著降低(>50%)、线粒体碎片化增加三倍和自噬增加 2.5 倍有关。在这些情况下,我们还注意到 PGC1α 的 mRNA 和蛋白丰度降低,以及关键线粒体成分(SDHA、ANT-1、UCP3 和 MFN2)的减少,以及细胞 ROS 增加和肌管中胰岛素作用受损。引人注目的是,NFkB 活性的药理学或遗传抑制改善了线粒体呼吸功能/形态的紊乱,减轻了 SDHA、ANT-1、UCP3 和 MFN2 的丧失,并减轻了 ROS 的增加和肌管胰岛素敏感性的降低。我们的研究结果表明,持续向骨骼肌细胞供应过多的代谢燃料会引起 NFkB 信号的增强,这是线粒体功能和形态、氧化还原状态和胰岛素信号紊乱的关键驱动因素。