Center for Genetic Epidemiology and Genomics, School of Public Health, Medical College of Soochow University, Suzhou, P. R. China.
Department of Epidemiology and Health Statistics, School of Public Health, Nantong University, Nantong, P. R. China.
J Cell Mol Med. 2019 Jul;23(7):4601-4610. doi: 10.1111/jcmm.14315. Epub 2019 May 20.
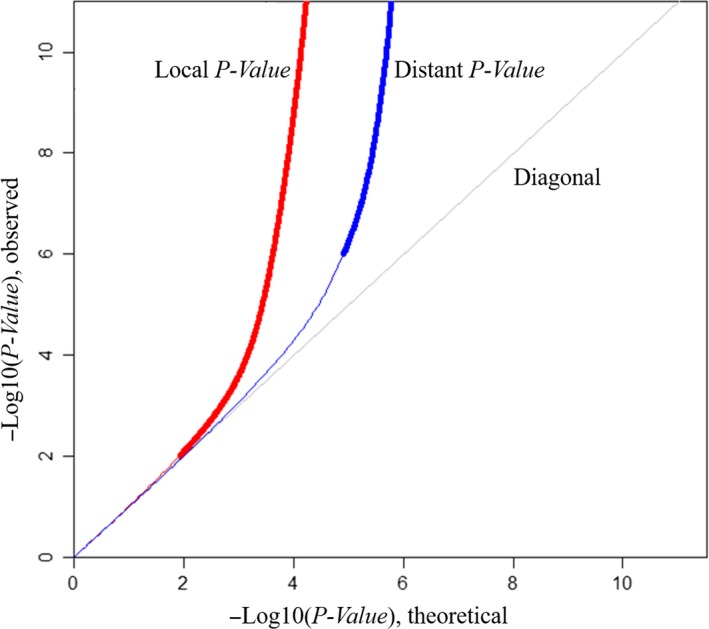
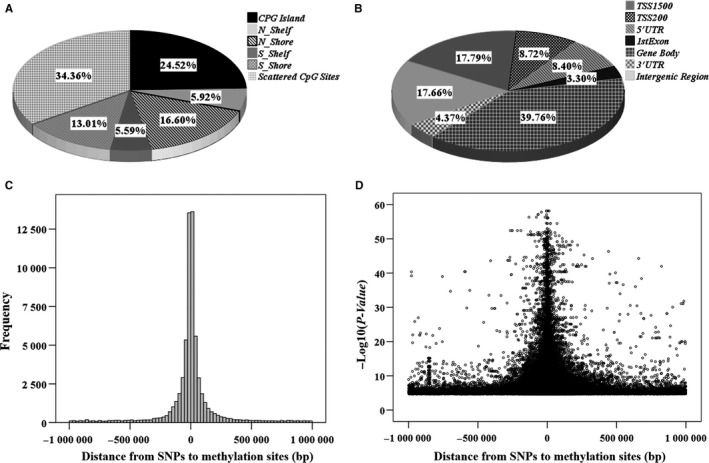
Genetic variants have potential influence on DNA methylation and thereby regulate mRNA expression. This study aimed to comprehensively reveal the relationships among SNP, methylation and mRNA, and identify methylation-mediated regulation patterns in human peripheral blood mononuclear cells (PBMCs). Based on in-house multi-omics datasets from 43 Chinese Han female subjects, genome-wide association trios were constructed by simultaneously testing the following three association pairs: SNP-methylation, methylation-mRNA and SNP-mRNA. Causal inference test (CIT) was used to identify methylation-mediated genetic effects on mRNA. A total of 64,184 significant cis-methylation quantitative trait loci (meQTLs) were identified (FDR < 0.05). Among the 745 constructed trios, 464 trios formed SNP-methylation-mRNA regulation chains (CIT). Network analysis (Cytoscape 3.3.0) constructed multiple complex regulation networks among SNP, methylation and mRNA (eg a total of 43 SNPs simultaneously connected to cg22517527 and further to PRMT2, DIP2A and YBEY). The regulation chains were supported by the evidence from 4DGenome database, relevant to immune or inflammatory related diseases/traits, and overlapped with previous eQTLs from dbGaP and GTEx. The results provide new insights into the regulation patterns among SNP, DNA methylation and mRNA expression, especially for the methylation-mediated effects, and also increase our understanding of functional mechanisms underlying the established associations.
遗传变异对 DNA 甲基化有潜在影响,从而调节 mRNA 的表达。本研究旨在全面揭示 SNP、甲基化和 mRNA 之间的关系,并确定人外周血单核细胞 (PBMCs) 中的甲基化介导的调控模式。基于 43 位中国汉族女性的内部多组学数据集,通过同时测试以下三个关联对,构建了全基因组关联三体:SNP-甲基化、甲基化-mRNA 和 SNP-mRNA。因果推理测试 (CIT) 用于识别甲基化对 mRNA 的遗传影响。共鉴定出 64,184 个显著的顺式-甲基化数量性状基因座 (meQTLs)(FDR<0.05)。在构建的 745 个三体中,464 个三体形成了 SNP-甲基化-mRNA 调控链 (CIT)。网络分析 (Cytoscape 3.3.0) 构建了 SNP、甲基化和 mRNA 之间的多个复杂调控网络(例如,共有 43 个 SNP 同时连接到 cg22517527,进一步连接到 PRMT2、DIP2A 和 YBEY)。这些调控链得到了 4DGenome 数据库的证据支持,与免疫或炎症相关疾病/特征相关,并且与 dbGaP 和 GTEx 中的先前 eQTL 重叠。研究结果为 SNP、DNA 甲基化和 mRNA 表达之间的调控模式提供了新的见解,特别是对于甲基化介导的效应,并且增加了我们对已建立关联背后的功能机制的理解。