Pathogen and Microbiome Institute, Northern Arizona University, Flagstaff, AZ 86011, USA.
Northern Arizona Healthcare, Flagstaff Medical Center, Flagstaff, AZ 86001, USA.
Microb Genom. 2019 Jul;5(7). doi: 10.1099/mgen.0.000271. Epub 2019 May 20.
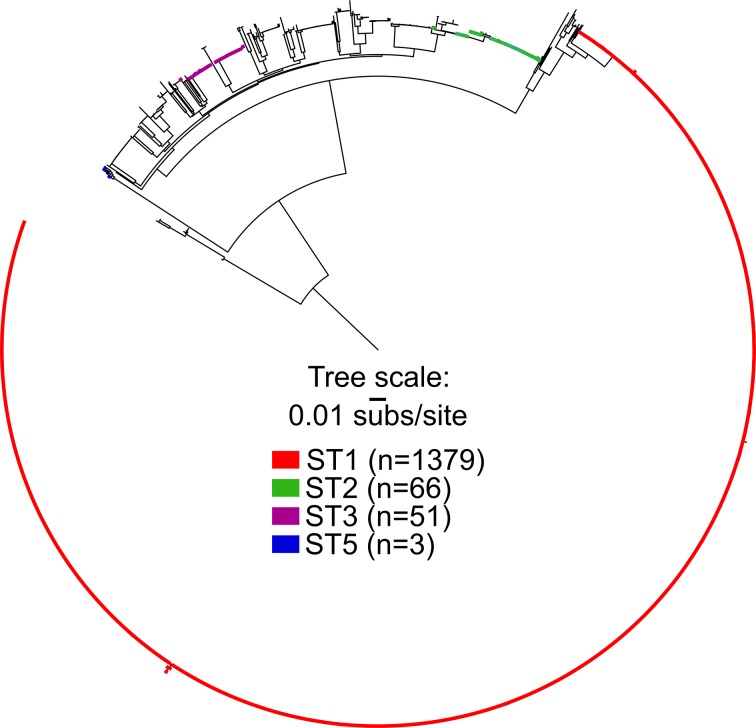
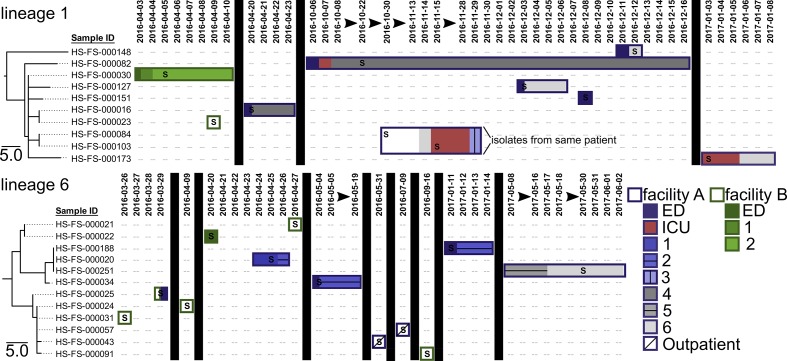
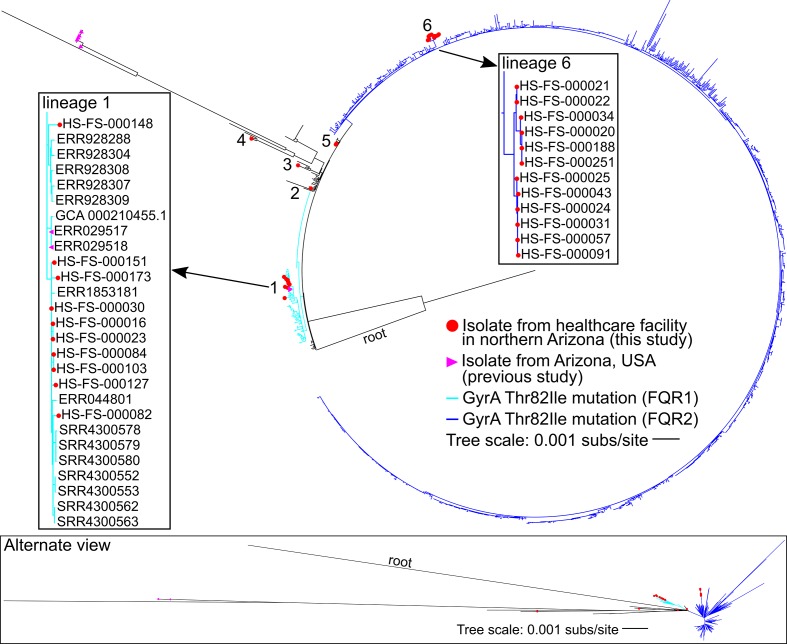
Clostridioides difficile is a ubiquitous, diarrhoeagenic pathogen often associated with healthcare-acquired infections that can cause a range of symptoms from mild, self-limiting disease to toxic megacolon and death. Since the early 2000s, a large proportion of C. difficile cases have been attributed to the ribotype 027 (RT027) lineage, which is associated with sequence type 1 (ST1) in the C. difficile multilocus sequence typing scheme. The spread of ST1 has been attributed, in part, to resistance to fluoroquinolones used to treat unrelated infections, which creates conditions ideal for C. difficile colonization and proliferation. In this study, we analysed 27 isolates from a healthcare network in northern Arizona, USA, and 1352 publicly available ST1 genomes to place locally sampled isolates into a global context. Whole genome, single nucleotide polymorphism analysis demonstrated that at least six separate introductions of ST1 were observed in healthcare facilities in northern Arizona over an 18-month sampling period. A reconstruction of transmission networks identified potential nosocomial transmission of isolates, which were only identified via whole genome sequence analysis. Antibiotic resistance heterogeneity was observed among ST1 genomes, including variability in resistance profiles among locally sampled ST1 isolates. To investigate why ST1 genomes are so common globally and in northern Arizona, we compared all high-quality C. difficile genomes and identified that ST1 genomes have gained and lost a number of genomic regions compared to all other C. difficile genomes; analyses of other toxigenic C. difficile sequence types demonstrate that this loss may be anomalous and could be related to niche specialization. These results suggest that a combination of antimicrobial resistance and gain and loss of specific genes may explain the prominent association of this sequence type with C. difficile infection cases worldwide. The degree of genetic variability in ST1 suggests that classifying all ST1 genomes into a quinolone-resistant hypervirulent clone category may not be appropriate. Whole genome sequencing of clinical C. difficile isolates provides a high-resolution surveillance strategy for monitoring persistence and transmission of C. difficile and for assessing the performance of infection prevention and control strategies.
艰难梭菌是一种普遍存在的、引起腹泻的病原体,常与医院获得性感染有关,可引起从轻度、自限性疾病到中毒性巨结肠和死亡等一系列症状。自 21 世纪初以来,很大一部分艰难梭菌病例归因于 027 型(RT027)谱系,该谱系与艰难梭菌多位点序列分型方案中的 1 型(ST1)有关。ST1 的传播部分归因于用于治疗无关感染的氟喹诺酮类药物的耐药性,这为艰难梭菌定植和增殖创造了理想的条件。在这项研究中,我们分析了来自美国亚利桑那州北部一个医疗保健网络的 27 个分离株和 1352 个公开的 ST1 基因组,将本地采样的分离株置于全球背景下。全基因组、单核苷酸多态性分析表明,在 18 个月的采样期间,亚利桑那州北部的医疗保健设施中至少观察到六次 ST1 的独立传入。传播网络的重建确定了分离株的潜在医院内传播,这些分离株仅通过全基因组序列分析才能识别。ST1 基因组中观察到抗生素耐药异质性,包括本地采样的 ST1 分离株之间耐药谱的差异。为了研究为什么 ST1 基因组在全球和亚利桑那州北部如此普遍,我们比较了所有高质量的艰难梭菌基因组,并发现与所有其他艰难梭菌基因组相比,ST1 基因组获得和丢失了许多基因组区域;对其他产毒艰难梭菌序列类型的分析表明,这种丢失可能是异常的,可能与生态位特化有关。这些结果表明,抗生素耐药性的获得和特定基因的获得和丢失的组合可能解释了该序列类型与全球艰难梭菌感染病例的显著关联。ST1 的遗传变异程度表明,将所有 ST1 基因组分类为喹诺酮耐药的高毒力克隆类别可能并不合适。临床艰难梭菌分离株的全基因组测序为监测艰难梭菌的持续存在和传播以及评估感染预防和控制策略的效果提供了一种高分辨率的监测策略。