Janezic Sandra, Rupnik Maja
National Laboratory for Health, Environment and Food, Maribor, Slovenia.
Medical Faculty, University of Maribor, Maribor, Slovenia.
Front Public Health. 2019 Oct 24;7:309. doi: 10.3389/fpubh.2019.00309. eCollection 2019.
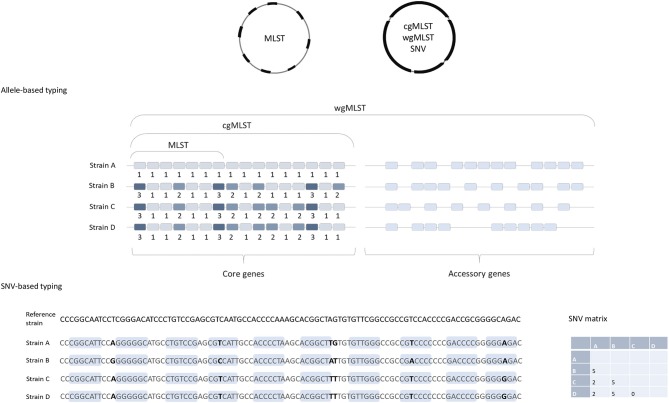
is an important nosocomial pathogen increasingly observed in the community and in different non-human reservoirs. The epidemiology and transmissibility of has been studied using a variety of typing methods, including more recently developed whole-genome sequence (WGS) analysis that is becoming used routinely for bacterial typing worldwide. Here we review the schemes for WGS-based typing methods available for and their applications in the field of human infection (CDI). The two main approaches to discover genomic variations are single nucleotide variant (SNV) analysis and methods based on gene-by-gene comparisons (frequently called core genome or whole genome MLST, cgMLST, or wgMLST). SNV analysis currently provides the ultimate resolution, however, typing nomenclature and standardized methodology are missing. On the other hand, gene-by-gene approaches allow portability and standardized nomenclature, and are therefore becoming increasingly popular in bacterial epidemiology and outbreak investigation. Two commercial software packages (BioNumerics and Ridom SeqSphere+) and an open source database (EnteroBase) for allele and sequence type determination for are currently available. Proof-of-concept WGS studies have already enabled advances in the investigation of the population structure of species, microevolution within the epidemic strains, intercontinental transmission over time and in tracking of transmission events. WGS of clinical isolates demonstrated a considerable genetic diversity suggesting diverse reservoirs for CDI. WGS was also shown to aid in resolving relapses and reinfections in recurrent CDI and has potential for use as a tool for assessing hospital infection prevention and control performance.
是一种重要的医院病原体,在社区和不同的非人类宿主中越来越多地被观察到。已经使用多种分型方法研究了其流行病学和传播性,包括最近开发的全基因组序列(WGS)分析,这种分析在全球范围内正逐渐常规用于细菌分型。在这里,我们综述了可用于的基于WGS的分型方法及其在人类感染(CDI)领域的应用。发现基因组变异的两种主要方法是单核苷酸变异(SNV)分析和基于逐个基因比较的方法(通常称为核心基因组或全基因组多位点序列分型,cgMLST,或wgMLST)。目前,SNV分析提供了最终的分辨率,然而,分型命名法和标准化方法尚不存在。另一方面,逐个基因的方法具有可移植性和标准化命名法,因此在细菌流行病学和疫情调查中越来越受欢迎。目前有两个用于确定等位基因和序列类型的商业软件包(BioNumerics和Ridom SeqSphere+)以及一个开源数据库(EnteroBase)。概念验证性的WGS研究已经在艰难梭菌种群结构调查、流行菌株内的微进化、随时间的洲际传播以及传播事件追踪方面取得了进展。临床艰难梭菌分离株的WGS显示出相当大的遗传多样性,表明CDI有多种宿主。WGS还被证明有助于解决复发性CDI中的复发和再感染问题,并且有潜力用作评估医院感染预防和控制绩效的工具。