Cardiometabolic Health Research School of Pharmacy & Life Sciences Robert Gordon University Aberdeen Scotland UK.
Present address: Institute of Cardiovascular & Medical Sciences College of Medical Veterinary and Life Sciences University of Glasgow Glasgow Scotland UK.
Pharmacol Res Perspect. 2019 May 24;7(3):e00487. doi: 10.1002/prp2.487. eCollection 2019 Jun.
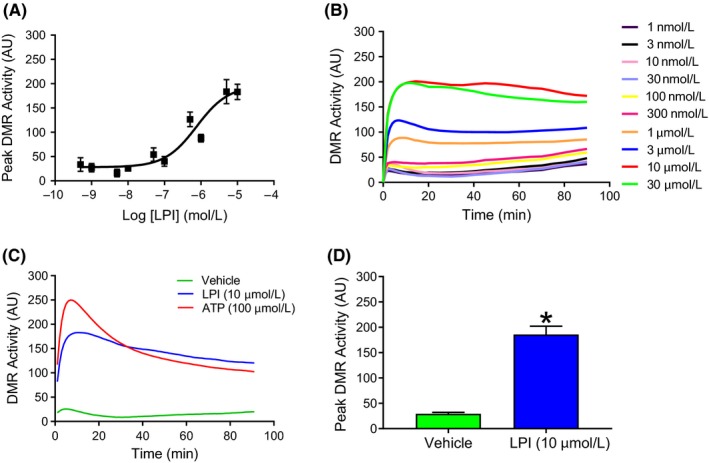
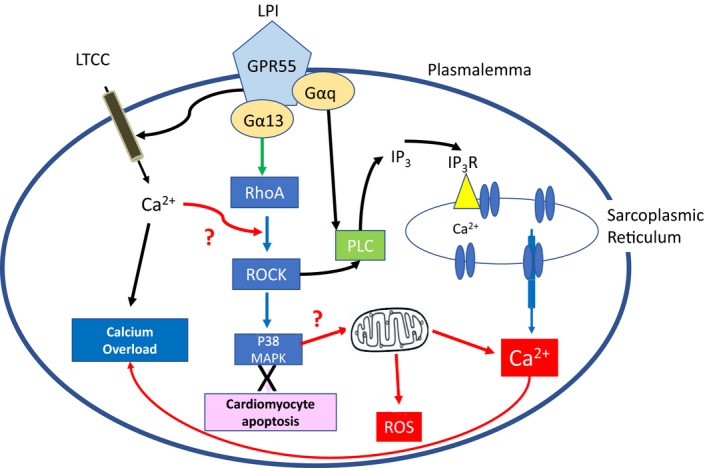
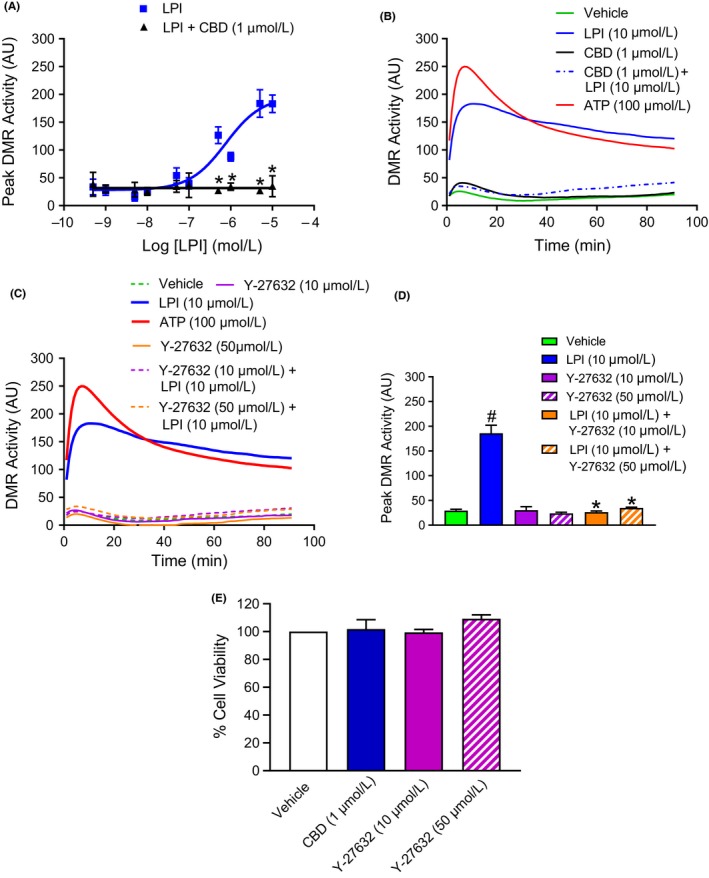
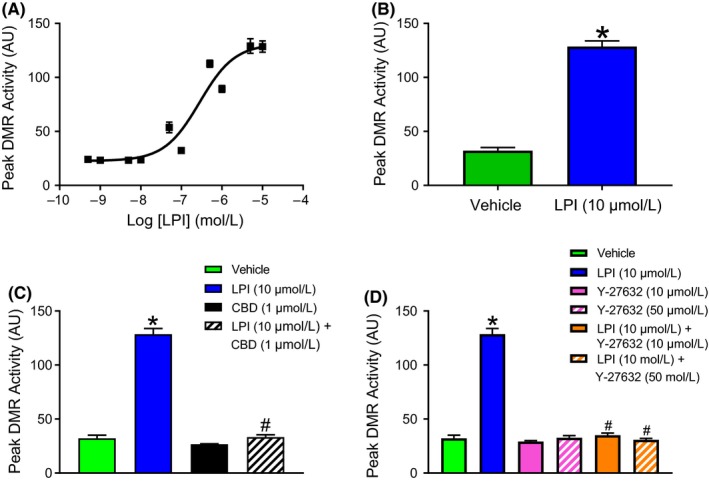
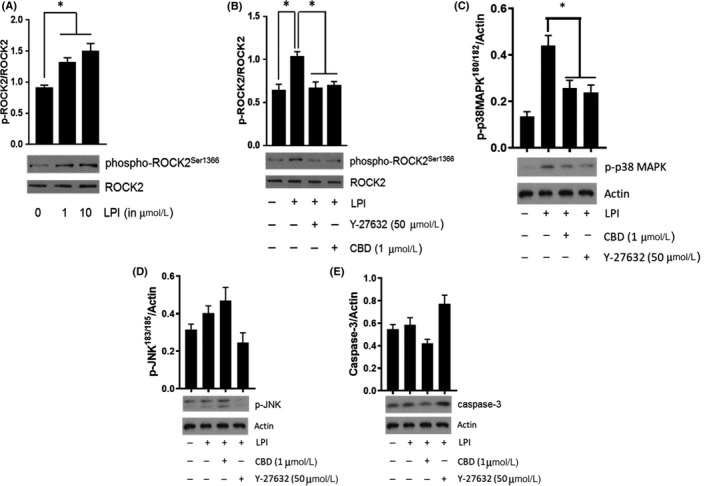
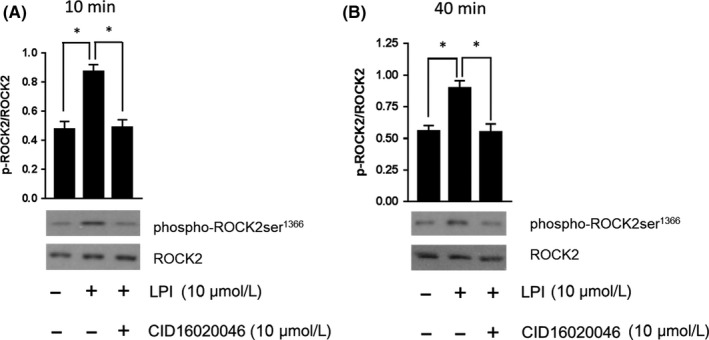
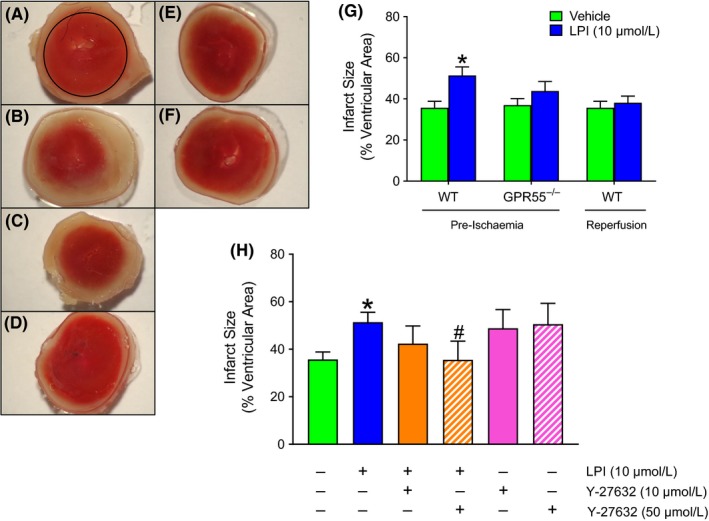
The phospholipid l-α-lysophosphatidylinositol (LPI), an endogenous ligand for GPR55, is elevated in patients with acute coronary syndrome, and a GPR55 antagonist cannabidiol (CBD) reduces experimental ischemia/reperfusion (I/R) injury. While LPI activates multiple signaling pathways, little is known about which ones are important in cardiomyocytes. In this study we explored whether activation of the Rho kinase/ROCK/p38 MAPK pathway is responsible for LPI-induced extension of I/R injury. Using a high-throughput screening method (dynamic mass redistribution; DMR), mouse- and human-induced pluripotent stem cell (iPSC) cardiomyocytes exposed to LPI were shown to exhibit a rapid, sustained, and concentration-dependent (1 nmol L-30 μmol L) cellular response. Y-27632 (ROCK inhibitor; 10 & 50 μmol L) and CBD (1 μmol L) both abolished the DMR response to LPI (10 μmol L). In murine iPSC cardiomyocytes, LPI-induced ROCK and p38 MAPK phosphorylation, both of which were prevented by Y-27632 and CBD, but did not induce JNK activation or cleavage of caspase-3. In hearts isolated from wild type (WT) mice subjected to 30 minutes global I/R, LPI (10 μmol L) administered via the coronary circulation increased infarct size when applied prior to ischemia onset, but not when given at the time of reperfusion. The exacerbation of tissue injury by LPI was not seen in hearts from GPR55 mice or in the presence of Y-27632, confirming that injury is mediated via the GPR55/ROCK/p38 MAPK pathway. These findings suggest that raised levels of LPI in the vicinity of a developing infarct may worsen the outcome of AMI.
磷脂酰肌醇 l-α-溶血磷脂酰肌醇(LPI)是 GPR55 的内源性配体,在急性冠状动脉综合征患者中升高,GPR55 拮抗剂大麻二酚(CBD)可减轻实验性缺血/再灌注(I/R)损伤。虽然 LPI 激活了多种信号通路,但对于哪些通路在心肌细胞中很重要知之甚少。在这项研究中,我们探讨了 Rho 激酶/ROCK/p38 MAPK 通路的激活是否负责 LPI 诱导的 I/R 损伤的扩展。使用高通量筛选方法(动态质量重分布;DMR),暴露于 LPI 的小鼠和人诱导多能干细胞(iPSC)心肌细胞表现出快速、持续和浓度依赖性(1 nmol L-30 μmol L)的细胞反应。Y-27632(ROCK 抑制剂;10 和 50 μmol L)和 CBD(1 μmol L)均消除了 LPI(10 μmol L)对 DMR 的反应。在鼠 iPSC 心肌细胞中,LPI 诱导 ROCK 和 p38 MAPK 磷酸化,这两种磷酸化均被 Y-27632 和 CBD 阻断,但不诱导 JNK 激活或半胱天冬酶-3 的裂解。在接受 30 分钟全脑 I/R 的野生型(WT)小鼠的心脏中,LPI(10 μmol L)通过冠状循环给药,在缺血发作前给药时可增加梗死面积,但在再灌注时给药则不会。在 GPR55 小鼠的心脏或在 Y-27632 存在的情况下,LPI 加剧组织损伤的现象并未出现,这证实损伤是通过 GPR55/ROCK/p38 MAPK 通路介导的。这些发现表明,在正在形成的梗死灶附近升高的 LPI 水平可能会使 AMI 的结果恶化。