Vascular Biology and Therapeutics Program (C.M.R., X.Z., N.R., B.A., J.R.K., V.U., Y.S., W.C.S., C.F.-H.), Yale University School of Medicine, New Haven, CT.
Integrative Cell Signaling and Neurobiology of Metabolism Program, Department of Comparative Medicine and Department of Pathology (C.M.R., X.Z., N.R., B.A., Y.S., C.F.-H.), Yale University School of Medicine, New Haven, CT.
Circulation. 2019 Jul 16;140(3):225-239. doi: 10.1161/CIRCULATIONAHA.118.038571. Epub 2019 Jun 3.
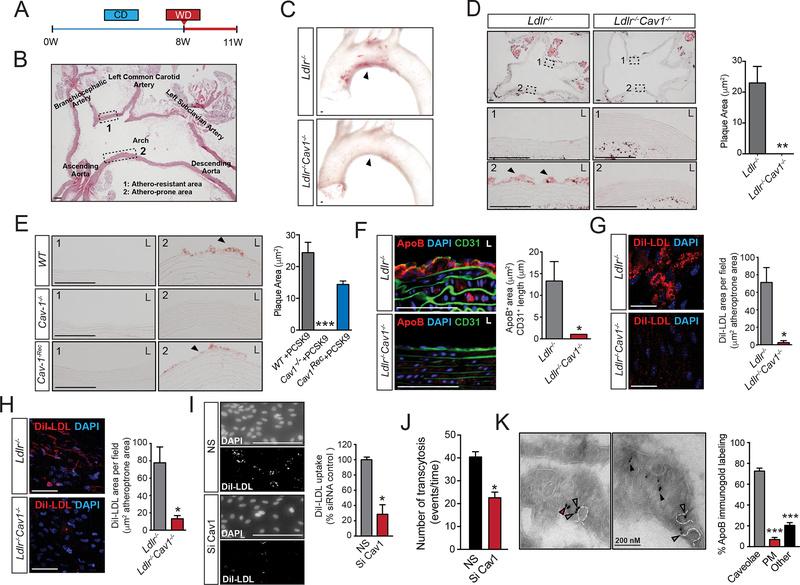
Atherosclerosis is driven by synergistic interactions between pathological, biomechanical, inflammatory, and lipid metabolic factors. Our previous studies demonstrated that absence of caveolin-1 (Cav1)/caveolae in hyperlipidemic mice strongly inhibits atherosclerosis, which was attributed to activation of endothelial nitric oxide (NO) synthase (eNOS) and increased production of NO and reduced inflammation and low-density lipoprotein trafficking. However, the contribution of eNOS activation and NO production in the athero-protection of Cav1 and the exact mechanisms by which Cav1/caveolae control the pathogenesis of diet-induced atherosclerosis are still not clear.
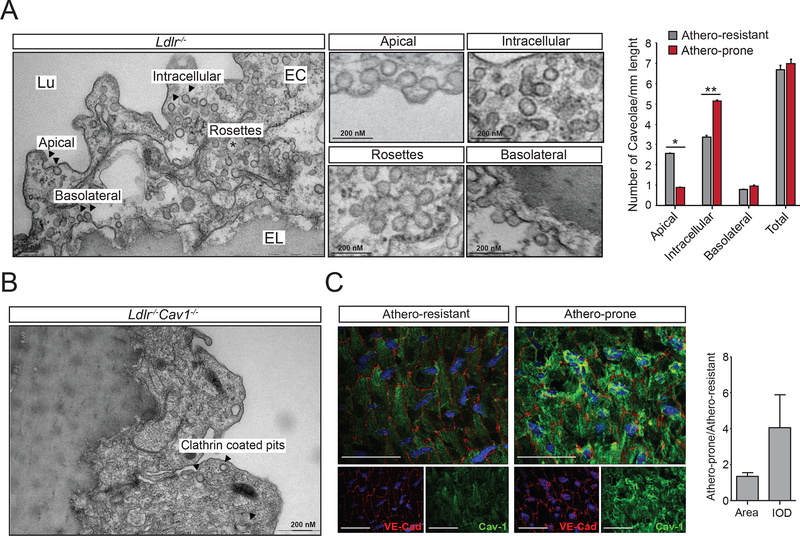
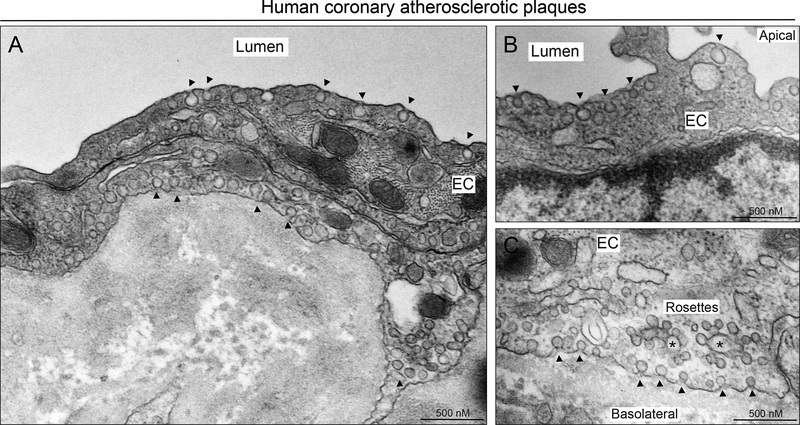
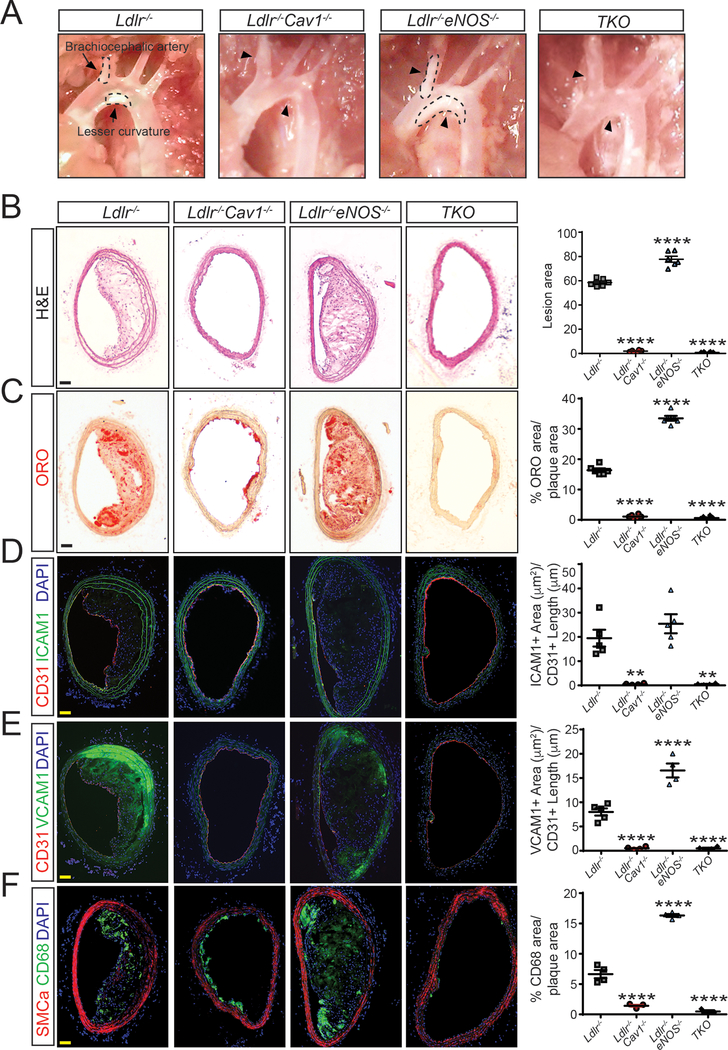
Triple-knockout mouse lacking expression of eNOS, Cav1, and Ldlr were generated to explore the role of NO production in Cav1-dependent athero-protective function. The effects of Cav1 on lipid trafficking, extracellular matrix remodeling, and vascular inflammation were studied both in vitro and in vivo with a mouse model of diet-induced atherosclerosis. The expression of Cav1 and distribution of caveolae regulated by flow were analyzed by immunofluorescence staining and transmission electron microscopy.
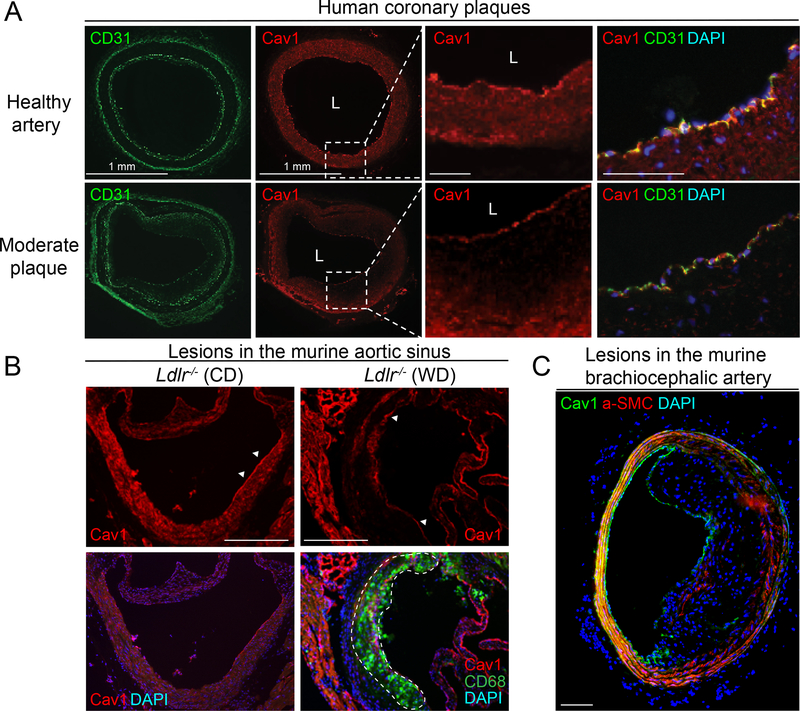
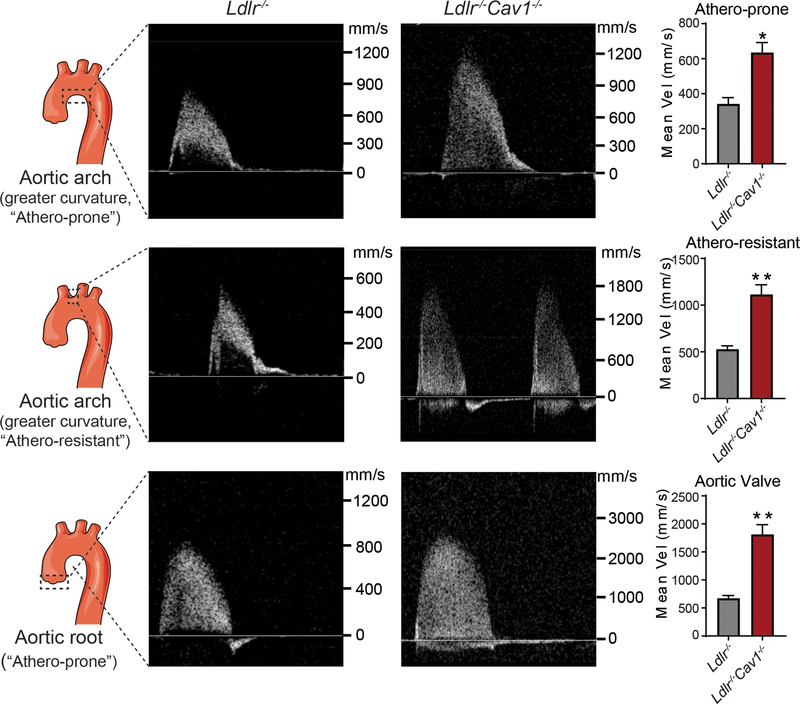
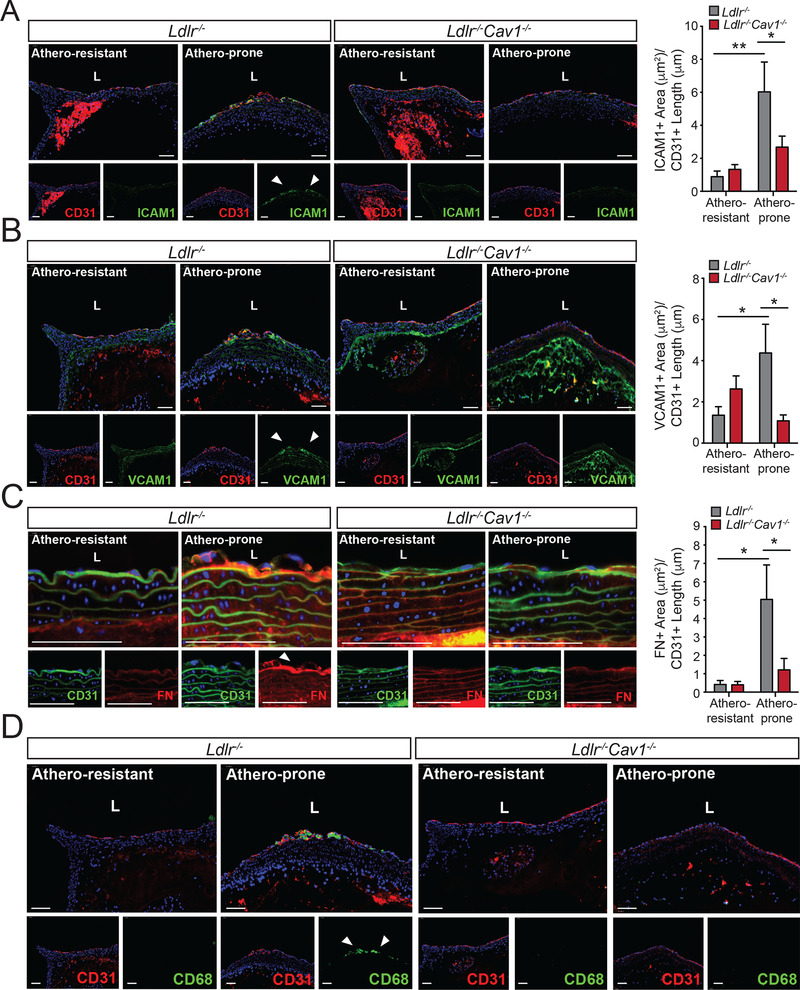
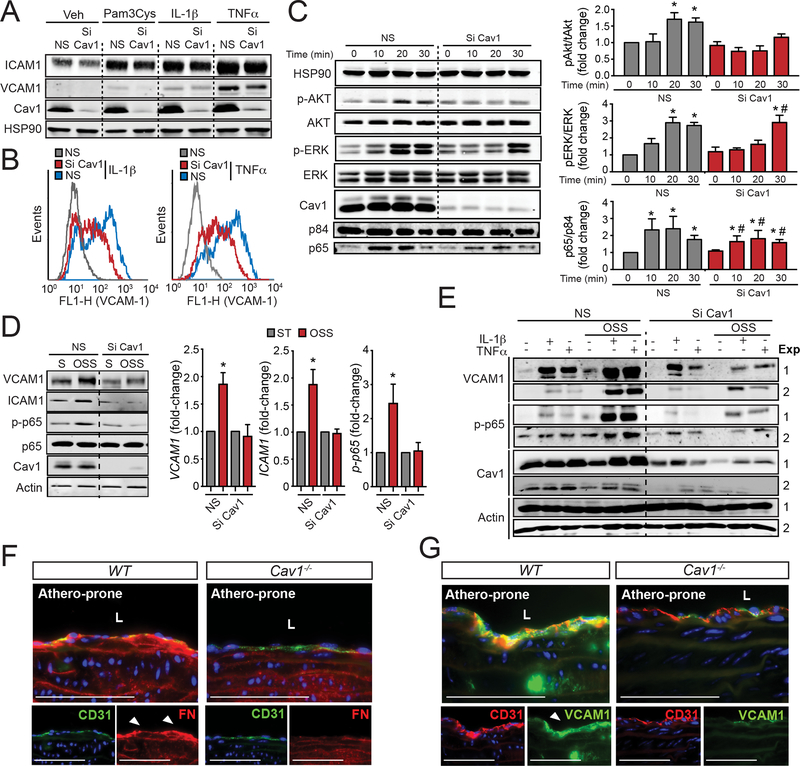
We found that absence of Cav1 significantly suppressed atherogenesis in LdlreNOS mice, demonstrating that athero-suppression is independent of increased NO production. Instead, we find that the absence of Cav1/caveolae inhibited low-density lipoprotein transport across the endothelium and proatherogenic fibronectin deposition and disturbed flow-mediated endothelial cell inflammation. Consistent with the idea that Cav1/caveolae may play a role in early flow-dependent inflammatory priming, distinct patterns of Cav1 expression and caveolae distribution were observed in athero-prone and athero-resistant areas of the aortic arch even in wild-type mice.
These findings support a role for Cav1/caveolae as a central regulator of atherosclerosis that links biomechanical, metabolic, and inflammatory pathways independently of endothelial eNOS activation and NO production.
动脉粥样硬化是由病理、生物力学、炎症和脂质代谢因素协同作用驱动的。我们之前的研究表明,高脂血症小鼠中 Cav1/小窝的缺失强烈抑制动脉粥样硬化,这归因于内皮型一氧化氮合酶(eNOS)的激活和一氧化氮(NO)的产生增加,以及炎症和低密度脂蛋白转运减少。然而,eNOS 激活和 NO 产生在 Cav1 保护动脉粥样硬化中的贡献以及 Cav1/小窝如何控制饮食诱导的动脉粥样硬化发病机制仍不清楚。
生成缺乏 eNOS、Cav1 和 Ldlr 表达的三重敲除小鼠,以探讨 NO 产生在 Cav1 依赖性动脉粥样硬化保护功能中的作用。利用饮食诱导的动脉粥样硬化小鼠模型,研究 Cav1 对脂质转运、细胞外基质重塑和血管炎症的影响。通过免疫荧光染色和透射电子显微镜分析 Cav1 的表达和由血流调节的小窝分布。
我们发现 Cav1 的缺失显著抑制了 LdlreNOS 小鼠的动脉粥样硬化形成,表明动脉粥样硬化抑制与增加的 NO 产生无关。相反,我们发现 Cav1/小窝的缺失抑制了低密度脂蛋白穿过内皮的转运以及促动脉粥样硬化的纤维连接蛋白沉积,并扰乱了血流介导的内皮细胞炎症。与 Cav1/小窝可能在早期血流依赖性炎症启动中发挥作用的观点一致,即使在野生型小鼠中,主动脉弓的动脉粥样硬化易损区和不易损区也观察到 Cav1 表达和小窝分布的不同模式。
这些发现支持 Cav1/小窝作为动脉粥样硬化的中央调节剂的作用,该作用独立于内皮型 eNOS 激活和 NO 产生,将生物力学、代谢和炎症途径联系起来。