Tao Shuxia, Schmidt Ines, Brocks Geert, Jiang Junke, Tranca Ionut, Meerholz Klaus, Olthof Selina
Center for Computational Energy Research, Department of Applied Physics, Eindhoven University of Technology, P.O. Box 513,, 5600MB, Eindhoven, The Netherlands.
Department of Chemistry, University of Cologne, Luxemburger Straße 116, Cologne, 50939, Germany.
Nat Commun. 2019 Jun 12;10(1):2560. doi: 10.1038/s41467-019-10468-7.
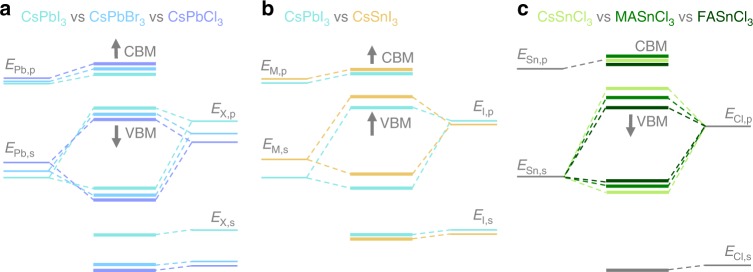
Metal halide perovskites are promising materials for future optoelectronic applications. One intriguing property, important for many applications, is the tunability of the band gap via compositional engineering. While experimental reports on changes in absorption or photoluminescence show rather good agreement for different compounds, the physical origins of these changes, namely the variations in valence and conduction band positions, are not well characterized. Here, we determine ionization energy and electron affinity values of all primary tin- and lead-based perovskites using photoelectron spectroscopy data, supported by first-principles calculations and a tight-binding analysis. We demonstrate energy level variations are primarily determined by the relative positions of the atomic energy levels of metal cations and halide anions and secondarily influenced by the cation-anion interaction strength. These results mark a significant step towards understanding the electronic structure of this material class and provides the basis for rational design rules regarding the energetics in perovskite optoelectronics.
金属卤化物钙钛矿是未来光电子应用中很有前景的材料。对于许多应用来说,一个有趣且重要的特性是通过成分工程实现带隙的可调性。虽然关于吸收或光致发光变化的实验报告表明不同化合物之间有相当好的一致性,但这些变化的物理起源,即价带和导带位置的变化,尚未得到很好的表征。在这里,我们利用光电子能谱数据,结合第一性原理计算和紧束缚分析,确定了所有主要的锡基和铅基钙钛矿的电离能和电子亲和能值。我们证明,能级变化主要由金属阳离子和卤化物阴离子的原子能级的相对位置决定,其次受阳离子 - 阴离子相互作用强度的影响。这些结果标志着在理解这类材料的电子结构方面迈出了重要一步,并为钙钛矿光电子学中能量学的合理设计规则提供了基础。