Soll Dominik, Spira Dominik, Hollstein Tim, Haberbosch Linus, Demuth Ilja, Steinhagen-Thiessen Elisabeth, Bobbert Thomas, Spranger Joachim, Kassner Ursula
Charité - Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin, Humboldt-Universität zu Berlin, Berlin Institute of Health, Lipid Clinic, Interdisciplinary Metabolism Center, Germany.
Berlin-Brandenburg Center for Regenerative Medicine (BCRT), Charité University Medicine Berlin, Germany.
Mol Genet Metab Rep. 2019 Jun 18;20:100479. doi: 10.1016/j.ymgmr.2019.100479. eCollection 2019 Sep.
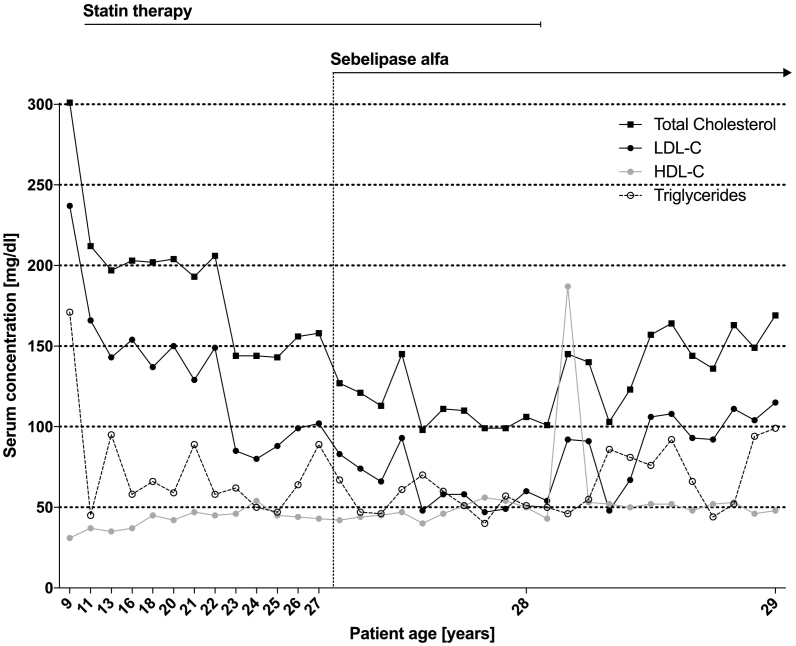
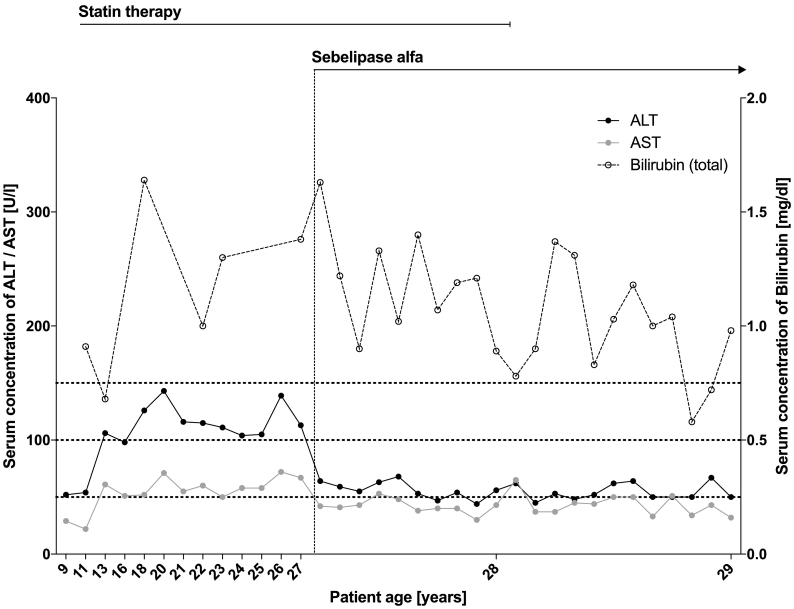
We report on a case of very rare autosomal recessive cholesteryl ester storage disease due to lysosomal acid lipase deficiency (LALD). LALD is caused by mutations in the lysosomal acid lipase A () gene resulting in cholesteryl ester accumulation in liver, spleen, and macrophages. It can lead to liver failure, accelerated atherosclerosis and premature death. Until recently, treatment options were limited to lipid-lowering medications to control dyslipidemia. Presently, a long-term enzyme replacement therapy with Sebelipase alfa, a recombinant human lysosomal acid lipase, is available for patients with LALD. Our patient's condition became conspicuous at the age of two due to a xanthogranuloma of the chin together with increased lipid levels, elevated liver enzymes and hepatomegaly. It took another five years until our patient was diagnosed with LALD after genetic testing. A bi-weekly therapy with intravenous Sebelipase alfa was started at the age of 26 years. It led to normalization of lipid levels, reduction of liver enzymes and beginning regression of hepatomegaly in the absence of adverse drug reactions after 46 infusions. Since LALD can take a fatal course even in patients with a long-term stable condition, it is essential to identify affected patients early and to treat them appropriately by enzyme replacement therapy. LALD should be suspected in patients with low high-density lipoprotein cholesterol (HDL-C) and high low-density lipoprotein cholesterol (LDL-C) in conjunction with elevated liver enzymes or hepatomegaly. A registry for LALD patients shall help to advance our understanding of the disease as well as improve patient care (NCT01633489).
我们报告了一例因溶酶体酸性脂肪酶缺乏(LALD)导致的极为罕见的常染色体隐性胆固醇酯贮积病病例。LALD由溶酶体酸性脂肪酶A()基因突变引起,导致胆固醇酯在肝脏、脾脏和巨噬细胞中蓄积。它可导致肝功能衰竭、加速动脉粥样硬化和过早死亡。直到最近,治疗选择还仅限于使用降血脂药物来控制血脂异常。目前,重组人溶酶体酸性脂肪酶塞贝脂肪酶α可用于LALD患者进行长期酶替代治疗。我们的患者在两岁时因下巴出现黄肉芽肿,同时血脂水平升高、肝酶升高和肝肿大而病情明显。又过了五年,我们的患者在经过基因检测后被诊断为LALD。在26岁时开始每两周静脉注射一次塞贝脂肪酶α进行治疗。在46次输注后,血脂水平恢复正常,肝酶降低,肝肿大开始消退,且未出现药物不良反应。由于即使是病情长期稳定的LALD患者也可能会有致命病程,因此早期识别受影响的患者并通过酶替代疗法进行适当治疗至关重要。对于高密度脂蛋白胆固醇(HDL-C)低、低密度脂蛋白胆固醇(LDL-C)高且伴有肝酶升高或肝肿大的患者,应怀疑患有LALD。一个LALD患者登记系统将有助于增进我们对该疾病的了解,并改善患者护理(NCT01633489)。