Wang Rui, Yang Zhongyi, Fei Yuhan, Feng Jiejie, Zhu Hui, Huang Fang, Zhang Hongsheng, Huang Ji
State Key Laboratory of Crop Genetics and Germplasm Enhancement, College of Agriculture, Nanjing Agricultural University, Nanjing, 210095, China.
BMC Genomics. 2019 Jun 28;20(1):534. doi: 10.1186/s12864-019-5879-7.
Usually the microRNA (miRNA)-mediated gene regulatory network (GRN) is constructed from the investigation of miRNA expression profiling and target predictions. However, the higher/lower expression level of miRNAs does not always indicate the higher/lower level of cleavages and such analysis, thus, sometimes ignores the crucial cleavage events. In the present work, the degradome sequencing data were employed to construct the complete miRNA-mediated gene regulatory network in soybean, unlike the traditional approach starting with small RNA sequencing data.
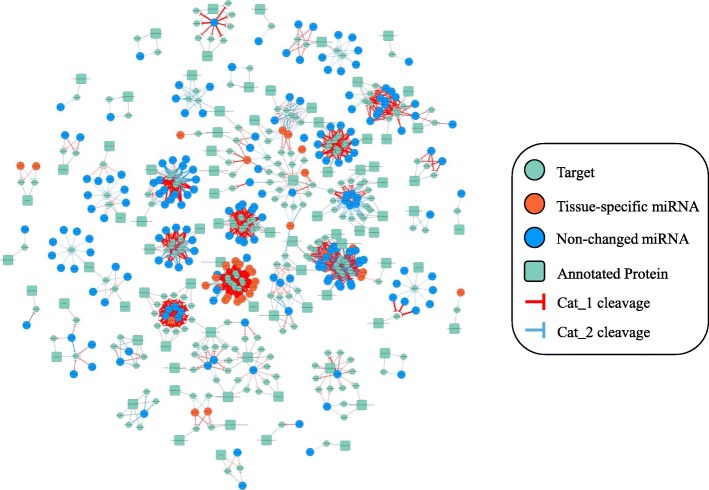
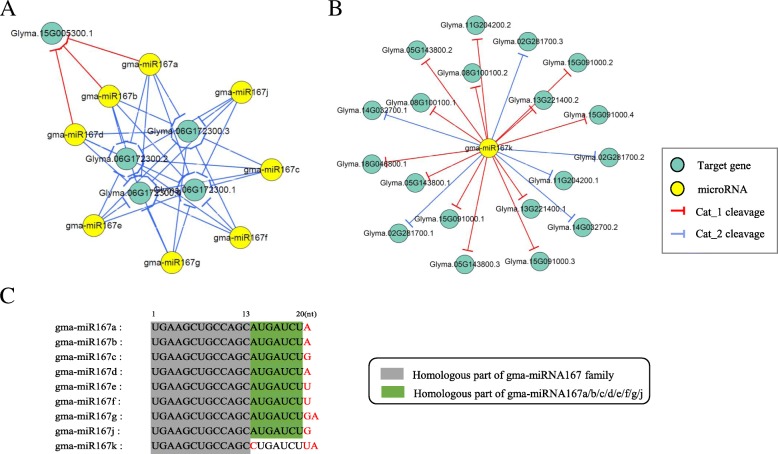
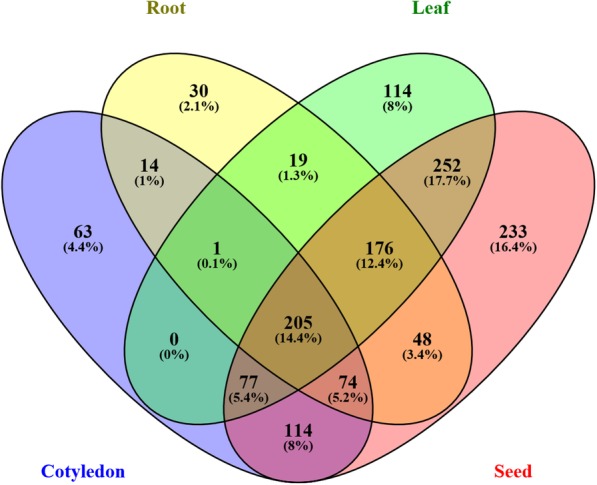
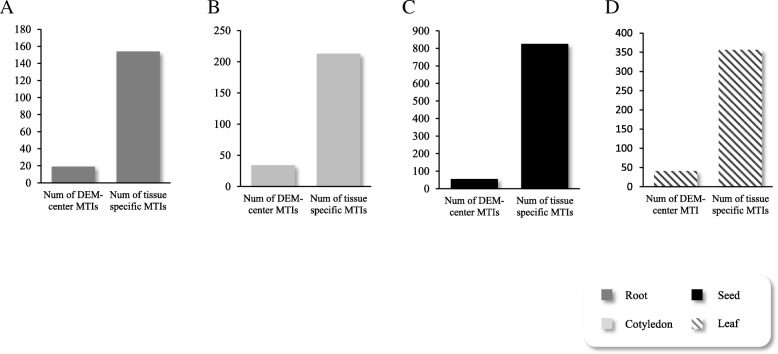
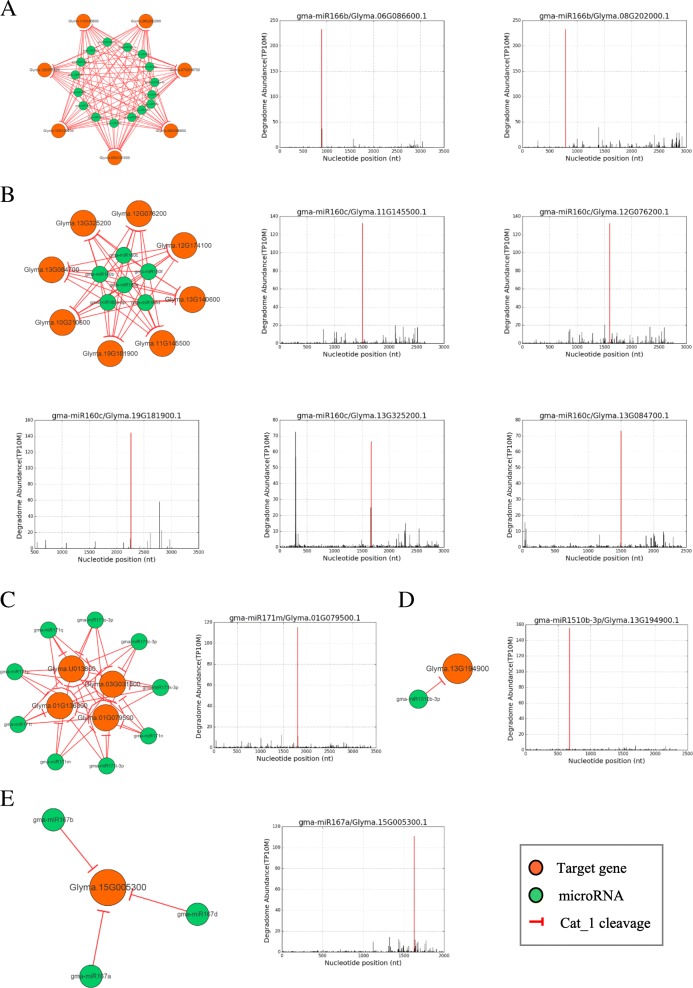
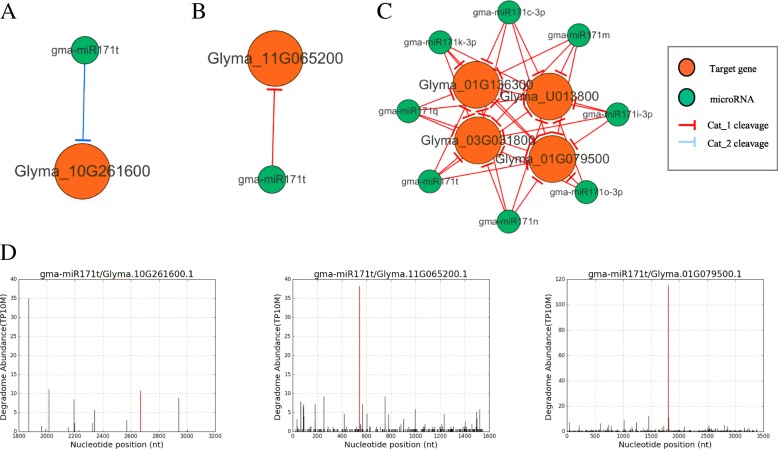
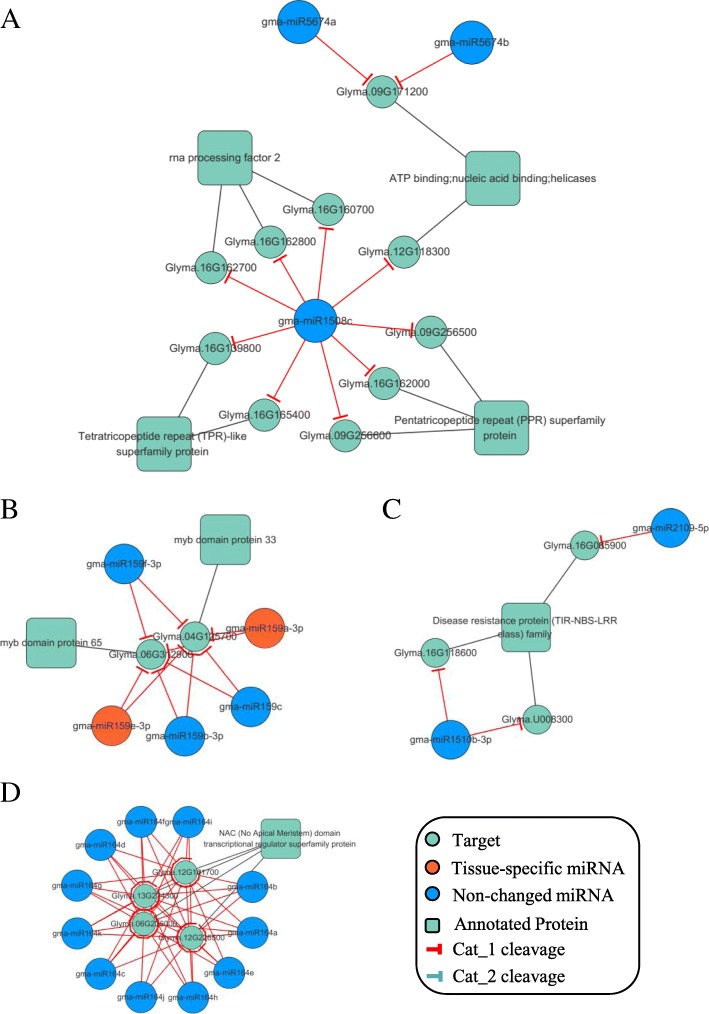
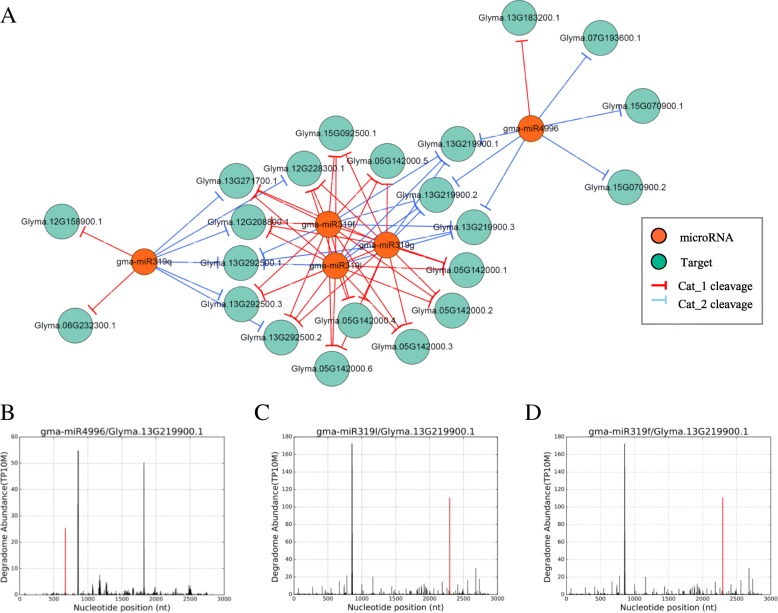
We constructed the root-, cotyledon-, leaf- and seed-specific miRNA regulatory networks with the degradome sequencing data and the forthcoming verification of miRNA profiling analysis. As a result, we identified 205 conserved miRNA-target interactions (MTIs) involved with 6 conserved gma-miRNA families and 365 tissue-specific MTIs containing 24 root-specific, 45 leaf-specific, 63 cotyledon-specific and 225 seed-specific MTIs. We found a total of 156 miRNAs in tissue-specific MTIs including 18 tissue-specific miRNAs, however, only 3 miRNAs have consistent tissue-specific expression. Our study showed the degradome-dependent miRNA regulatory networks (DDNs) in four soybean tissues and explored their conservations and specificities.
The construction of DDNs may provide the complete miRNA-Target interactions in certain plant tissues, leading to the identification of the conserved and tissue-specific MTIs and sub-networks. Our work provides a basis for further investigation of the roles and mechanisms of miRNA-mediated regulation of tissue-specific growth and development in soybean.
通常,微小RNA(miRNA)介导的基因调控网络(GRN)是通过对miRNA表达谱和靶标预测的研究构建的。然而,miRNA的表达水平升高/降低并不总是意味着切割水平的升高/降低,因此,这种分析有时会忽略关键的切割事件。在本研究中,我们利用降解组测序数据构建了大豆中完整的miRNA介导的基因调控网络,这与传统的从小RNA测序数据开始的方法不同。
我们利用降解组测序数据以及即将进行的miRNA谱分析验证,构建了根、子叶、叶和种子特异性的miRNA调控网络。结果,我们鉴定出205个保守的miRNA-靶标相互作用(MTIs),涉及6个保守的gma-miRNA家族,以及365个组织特异性的MTIs,其中包括24个根特异性、45个叶特异性、63个子叶特异性和225个种子特异性的MTIs。我们在组织特异性MTIs中总共发现了156个miRNA,其中包括18个组织特异性miRNA,然而,只有3个miRNA具有一致的组织特异性表达。我们的研究展示了大豆四个组织中依赖降解组的miRNA调控网络(DDNs),并探索了它们的保守性和特异性。
DDNs的构建可以提供特定植物组织中完整的miRNA-靶标相互作用,从而鉴定出保守的和组织特异性的MTIs以及子网络。我们的工作为进一步研究miRNA介导的大豆组织特异性生长和发育的作用及机制提供了基础。