Department of Chemical and Biological Engineering, Colorado State University, Fort Collins, CO, United States of America.
Bioscience Division, Los Alamos National Laboratory, Los Alamos, NM, United States of America.
PLoS One. 2019 Jul 1;14(7):e0215502. doi: 10.1371/journal.pone.0215502. eCollection 2019.
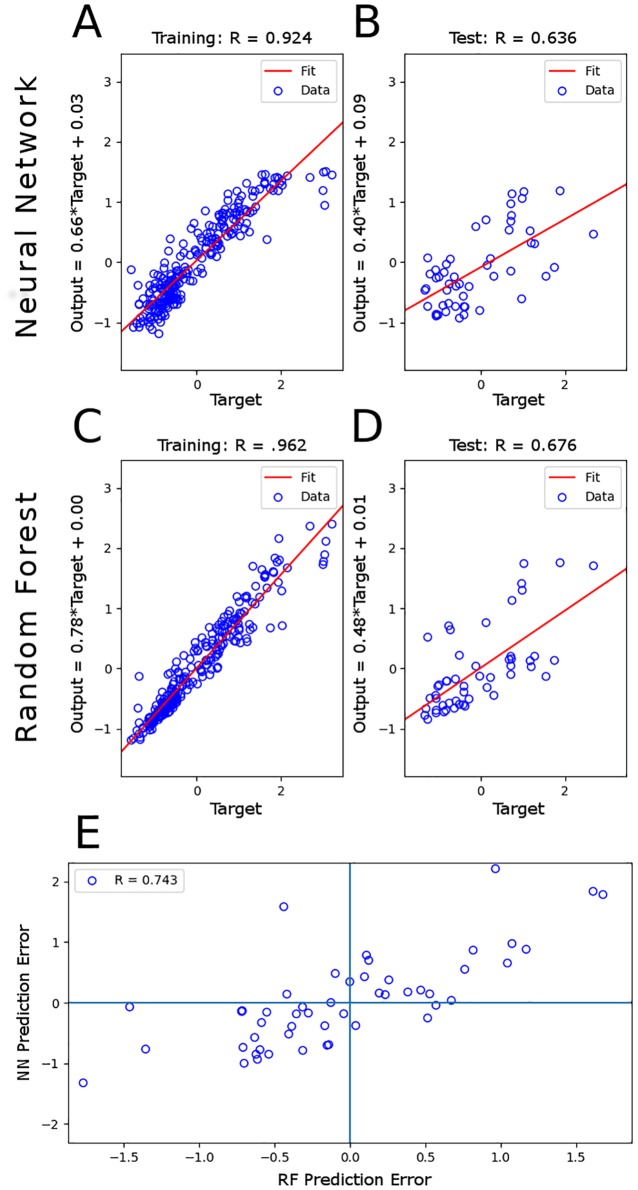
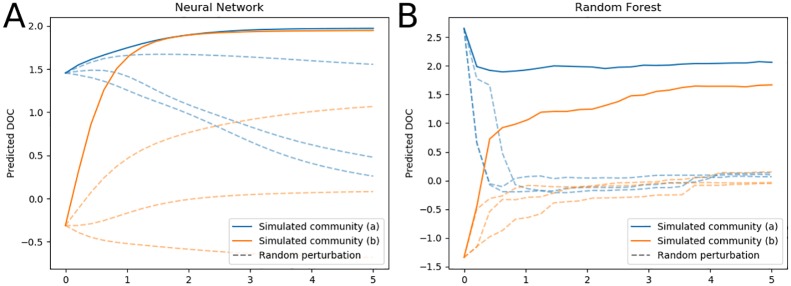
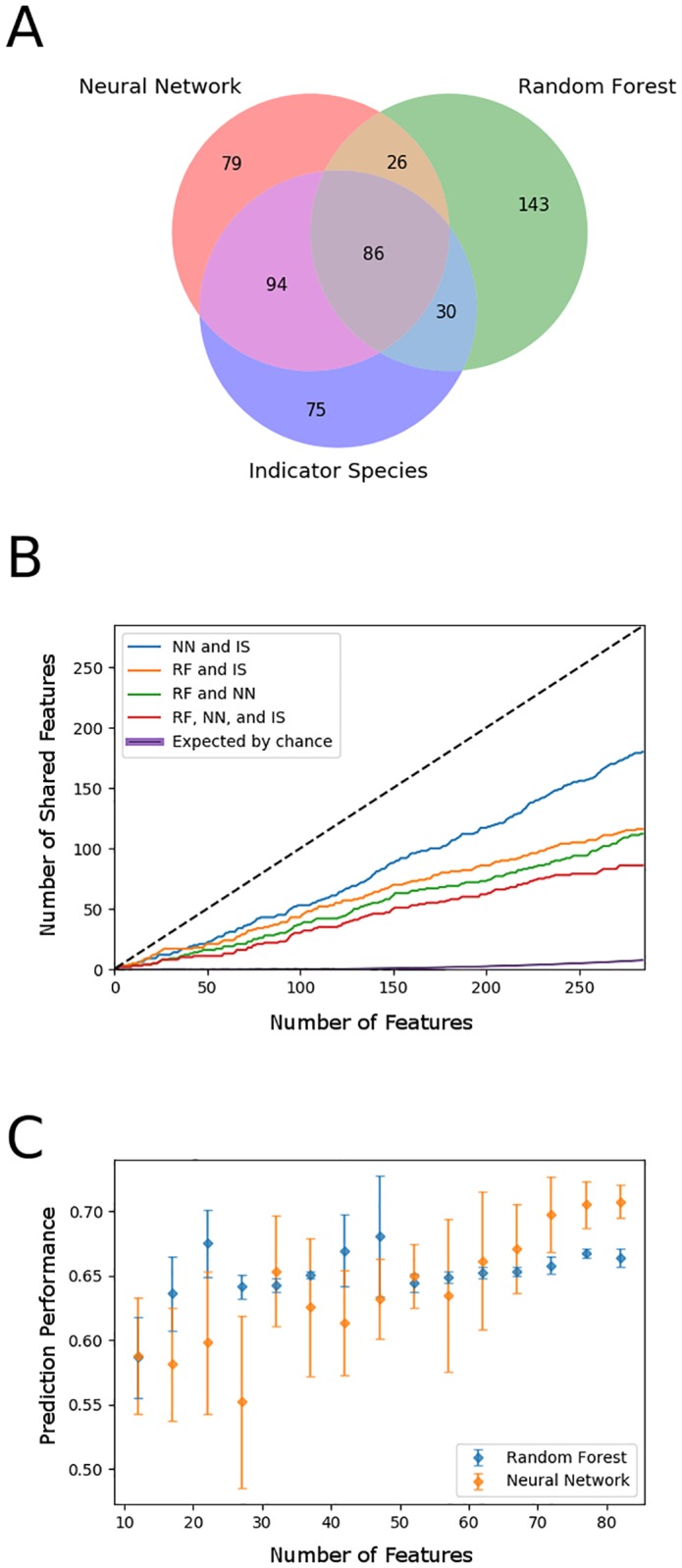
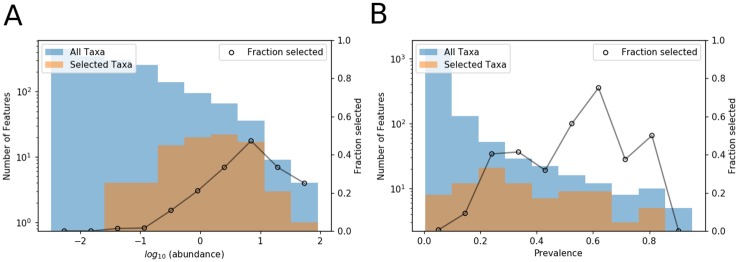
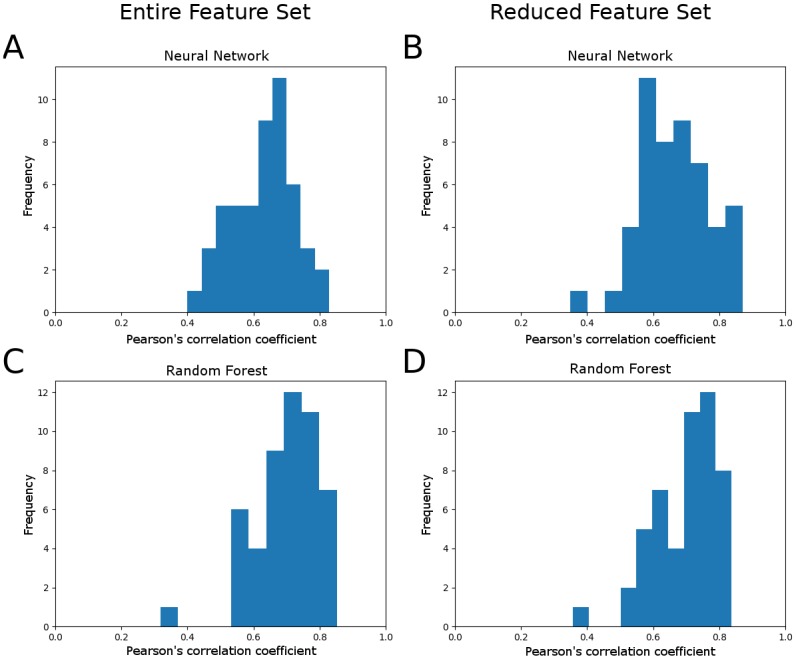
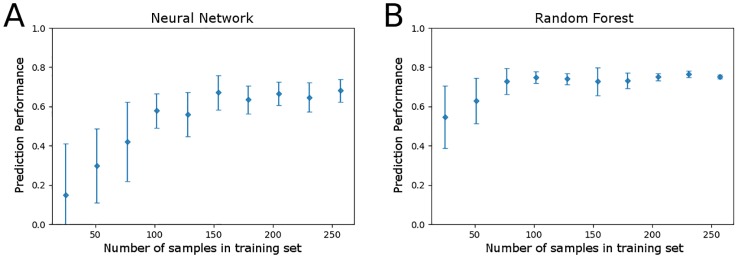
Microbial communities are ubiquitous and often influence macroscopic properties of the ecosystems they inhabit. However, deciphering the functional relationship between specific microbes and ecosystem properties is an ongoing challenge owing to the complexity of the communities. This challenge can be addressed, in part, by integrating the advances in DNA sequencing technology with computational approaches like machine learning. Although machine learning techniques have been applied to microbiome data, use of these techniques remains rare, and user-friendly platforms to implement such techniques are not widely available. We developed a tool that implements neural network and random forest models to perform regression and feature selection tasks on microbiome data. In this study, we applied the tool to analyze soil microbiome (16S rRNA gene profiles) and dissolved organic carbon (DOC) data from a 44-day plant litter decomposition experiment. The microbiome data includes 1709 total bacterial operational taxonomic units (OTU) from 300+ microcosms. Regression analysis of predicted and actual DOC for a held-out test set of 51 samples yield Pearson's correlation coefficients of.636 and.676 for neural network and random forest approaches, respectively. Important taxa identified by the machine learning techniques are compared to results from a standard tool (indicator species analysis) widely used by microbial ecologists. Of 1709 bacterial taxa, indicator species analysis identified 285 taxa as significant determinants of DOC concentration. Of the top 285 ranked features determined by machine learning methods, a subset of 86 taxa are common to all feature selection techniques. Using this subset of features, prediction results for random permutations of the data set are at least equally accurate compared to predictions determined using the entire feature set. Our results suggest that integration of multiple methods can aid identification of a robust subset of taxa within complex communities that may drive specific functional outcomes of interest.
微生物群落无处不在,通常会影响它们所栖息的生态系统的宏观特性。然而,由于群落的复杂性,破译特定微生物与生态系统特性之间的功能关系仍然是一个持续的挑战。这个挑战可以部分通过将 DNA 测序技术的进步与机器学习等计算方法相结合来解决。尽管机器学习技术已应用于微生物组数据,但这些技术的使用仍然很少,并且没有广泛可用的用户友好型平台来实施这些技术。我们开发了一种工具,该工具实现了神经网络和随机森林模型,可对微生物组数据执行回归和特征选择任务。在这项研究中,我们应用该工具来分析来自 44 天植物凋落叶分解实验的土壤微生物组(16S rRNA 基因谱)和溶解有机碳(DOC)数据。微生物组数据包括来自 300 多个微宇宙的 1709 个总细菌操作分类单元(OTU)。对 51 个样本的保留测试集的预测和实际 DOC 的回归分析,神经网络和随机森林方法的 Pearson 相关系数分别为 0.636 和 0.676。机器学习技术确定的重要分类群与微生物生态学家广泛使用的标准工具(指示物种分析)的结果进行了比较。在 1709 个细菌分类群中,指示物种分析确定了 285 个分类群是 DOC 浓度的重要决定因素。在机器学习方法确定的前 285 个排名特征中,有 86 个分类群是所有特征选择技术共有的。使用该特征子集,与使用整个特征集确定的预测结果相比,对数据集的随机排列进行的预测结果至少同样准确。我们的结果表明,整合多种方法可以帮助识别复杂群落中可能驱动特定感兴趣功能结果的稳健分类群子集。