Instituto de Investigaciones Biomédicas "Alberto Sols", Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid (CSIC-UAM), C/ Arturo Duperier, 4, 28029, Madrid, Spain.
Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Instituto de Salud Carlos III, C/ Valderrebollo, 5, 28031, Madrid, Spain.
Cell Death Dis. 2019 Jul 11;10(7):535. doi: 10.1038/s41419-019-1766-z.
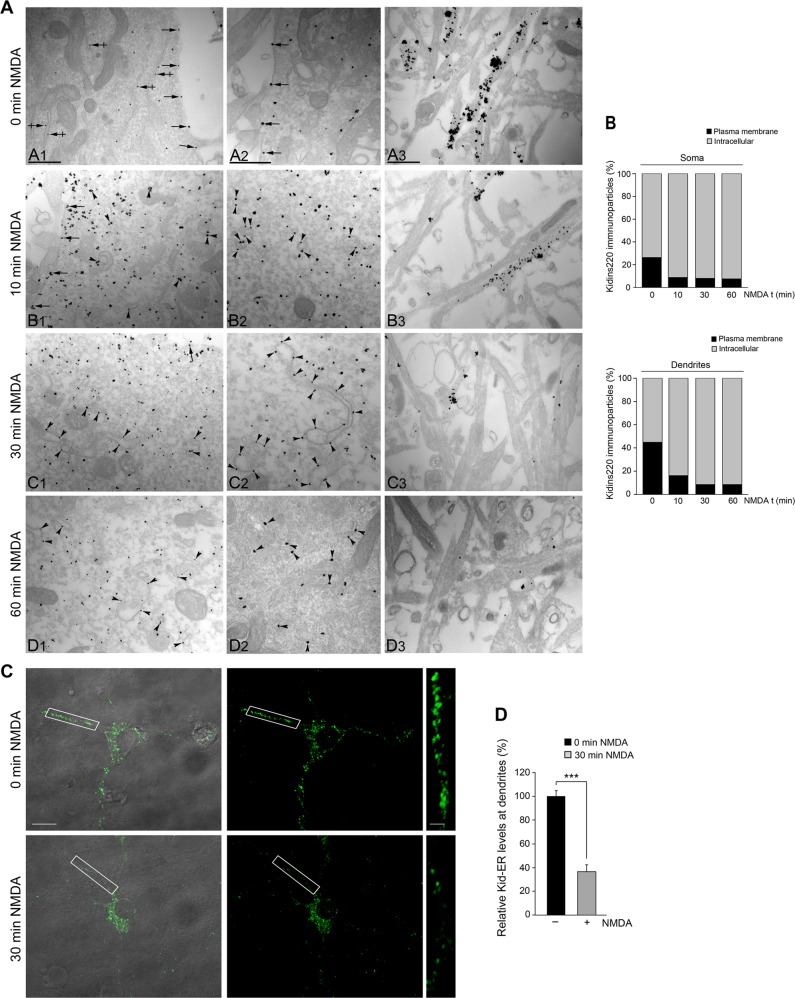
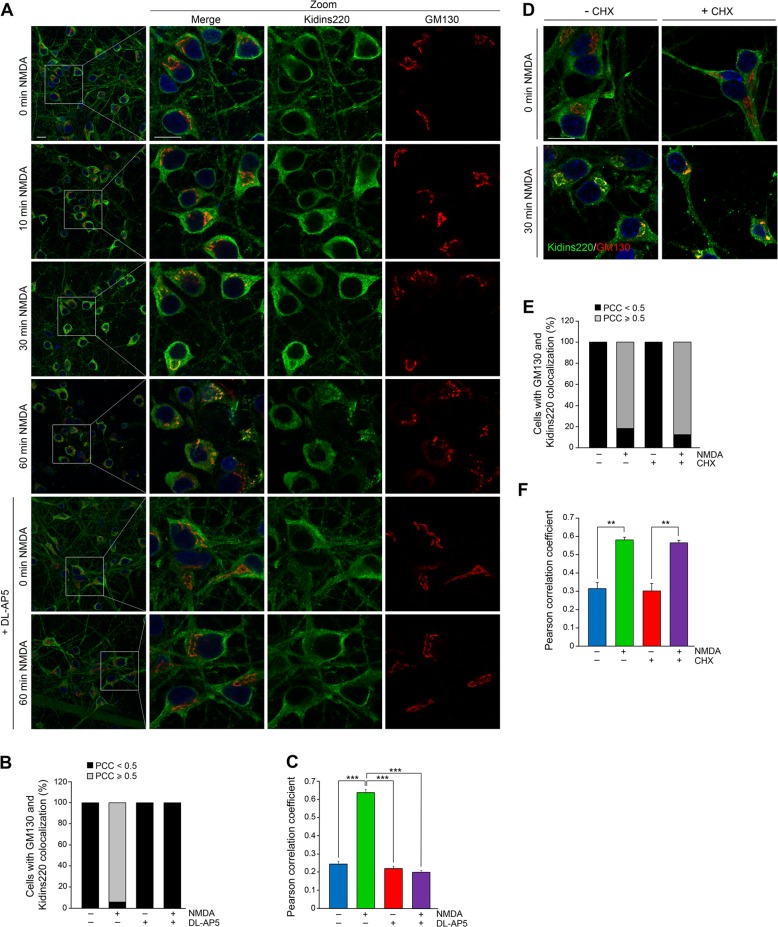
Excitotoxic neuronal death induced by high concentrations of glutamate is a pathological event common to multiple acute or chronic neurodegenerative diseases. Excitotoxicity is mediated through overactivation of the N-Methyl-D-aspartate type of ionotropic glutamate receptors (NMDARs). Physiological stimulation of NMDARs triggers their endocytosis from the neuronal surface, inducing synaptic activity and survival. However almost nothing is known about the internalization of overactivated NMDARs and their interacting proteins, and how this endocytic process is connected with neuronal death has been poorly explored. Kinase D-interacting substrate of 220 kDa (Kidins220), also known as ankyrin repeat-rich membrane spanning (ARMS), is a component of NMDAR complexes essential for neuronal viability by the control of ERK activation. Here we have investigated Kidins220 endocytosis induced by NMDAR overstimulation and the participation of this internalization step in the molecular mechanisms of excitotoxicity. We show that excitotoxicity induces Kidins220 and GluN1 traffic to the Golgi apparatus (GA) before Kidins220 is degraded by the protease calpain. We also find that excitotoxicity triggers an early activation of Rap1-GTPase followed by its inactivation. Kidins220 excitotoxic endocytosis and subsequent calpain-mediated downregulation governs this late inactivation of Rap1 that is associated to decreases in ERK activity preceding neuronal death. Furthermore, we identify the molecular mechanisms involved in the excitotoxic shutoff of Kidins220/Rap1/ERK prosurvival cascade that depends on calpain processing of Rap1-activation complexes. Our data fit in a model where Kidins220 targeting to the GA during early excitotoxicity would facilitate Rap1 activation and subsequent stimulation of ERK. At later times, activation of Golgi-associated calpain, would promote the degradation of GA-targeted Kidins220 and two additional components of the specific Rap1 activation complex, PDZ-GEF1, and S-SCAM. In this way, late excitotoxicity would turn off Rap1/ERK cascade and compromise neuronal survival.
由高浓度谷氨酸诱导的兴奋性神经元死亡是多种急性或慢性神经退行性疾病的共同病理事件。兴奋性毒性是通过离子型谷氨酸受体(NMDARs)的过度激活介导的。NMDAR 的生理刺激会触发其从神经元表面内化,诱导突触活动和存活。然而,对于过度激活的 NMDAR 及其相互作用蛋白的内化以及该内化过程如何与神经元死亡相关,我们知之甚少。激酶 D 相互作用的 220kDa 底物(Kidins220),也称为富含锚蛋白重复的跨膜(ARMS),是 NMDAR 复合物的一个组成部分,通过控制 ERK 激活对神经元存活至关重要。在这里,我们研究了 NMDAR 过度刺激诱导的 Kidins220 内吞作用以及该内化步骤在兴奋性毒性的分子机制中的参与。我们发现,兴奋性毒性会诱导 Kidins220 和 GluN1 向高尔基体(GA)运输,然后 Kidins220 被蛋白酶钙蛋白酶降解。我们还发现,兴奋性毒性会触发 Rap1-GTPase 的早期激活,随后其失活。Kidins220 的兴奋性内吞作用和随后的钙蛋白酶介导的下调控制了 Rap1 的晚期失活,这与神经元死亡前 ERK 活性的降低有关。此外,我们确定了参与 Kidins220/Rap1/ERK 促生存级联反应的兴奋性关闭的分子机制,该机制依赖于钙蛋白酶对 Rap1 激活复合物的处理。我们的数据符合这样一种模式,即在早期兴奋性毒性中,Kidins220 向 GA 的靶向作用将促进 Rap1 的激活,随后刺激 ERK。在稍后的时间点,高尔基体相关钙蛋白酶的激活将促进 GA 靶向的 Kidins220 和特定 Rap1 激活复合物的另外两个组成部分 PDZ-GEF1 和 S-SCAM 的降解。通过这种方式,晚期兴奋性毒性会关闭 Rap1/ERK 级联反应并损害神经元存活。