Department of Hygiene, Epidemiology and Medical Statistics, Medical School, National and Kapodistrian University of Athens, Athens, Greece.
Department of Biological Sciences, University of Cyprus, Nicosia, Cyprus.
Sci Rep. 2019 Jul 11;9(1):10077. doi: 10.1038/s41598-019-46552-7.
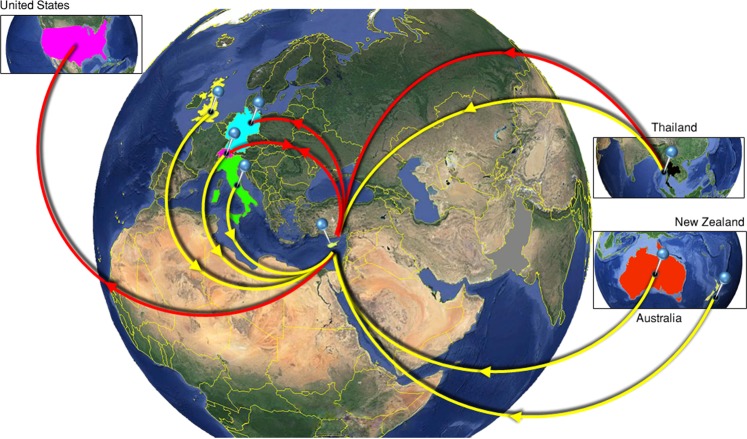
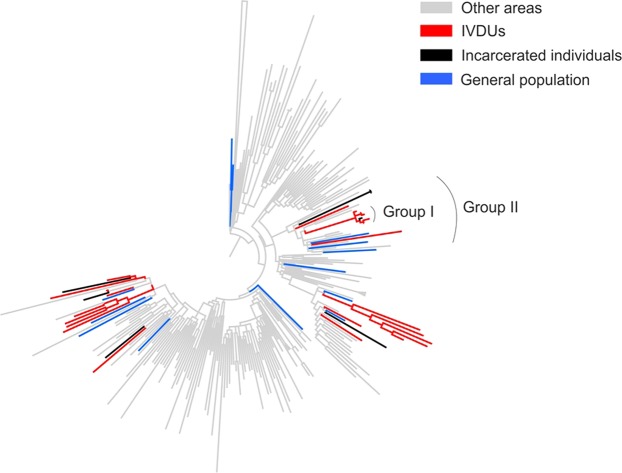
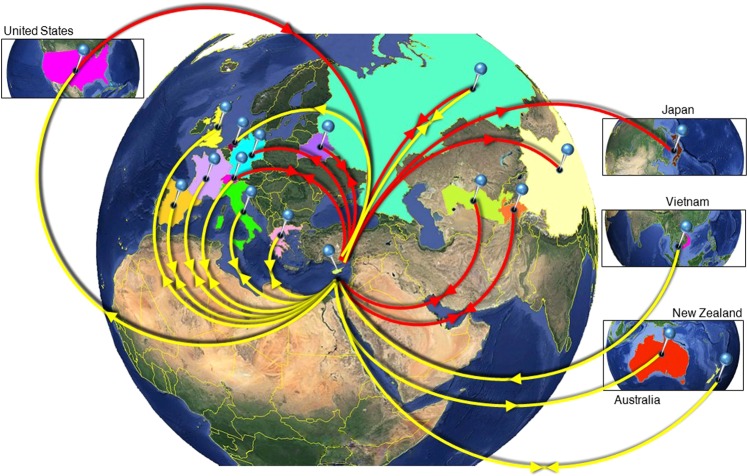
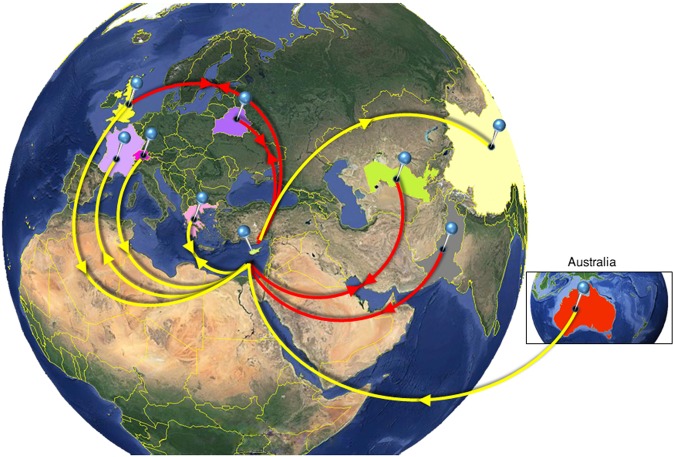
Hepatitis C virus (HCV) genotype and subtype distribution differs according to geographic origin and transmission risk category. Previous molecular epidemiology studies suggest the presence of multiple subtypes among Cypriot subjects. To investigate HCV genotype- and subtype-specific dissemination patterns, origins, and transmission in Cyprus, we analyzed HCV sequences encoding partial Core-E1 and NS5B regions. Analyzed populations comprised the general population and high-risk cohorts in Cyprus and a globally sampled dataset. Maximum-likelihood phylogeny reconstruction with bootstrap evaluation, character reconstruction using parsimony, and bootstrap trees estimated by ML were performed to identify the geographic origin of HCV subtypes and statistically significant dispersal pathways among geographic regions. Phylogeographic analyses traced the origin of subtypes in the general population and among PWID in Cyprus to unique and overlapping globally distributed regions. Phylogenetic analysis in Core-E1 revealed that most sequences from incarcerated populations in Cyprus clustered with the general population and PWID. We estimate that HCV infections in Cyprus originate from multiple global sources while most HCV transmissions among incarcerated individuals occur locally. This analysis is one of a few studies tracing HCV dispersal patterns using global datasets, and these practices and findings should inform how HCV epidemics are targeted by future prevention policies.
丙型肝炎病毒(HCV)基因型和亚型的分布因地理起源和传播风险类别而异。先前的分子流行病学研究表明,塞浦路斯人群中存在多种亚型。为了调查塞浦路斯 HCV 基因型和亚型特异性的传播模式、起源和传播,我们分析了 HCV 编码部分核心-E1 和 NS5B 区的序列。分析人群包括塞浦路斯的一般人群和高危人群以及全球抽样数据集。使用最大似然法进行系统发育重建,并进行 bootstrap 评估,使用简约法进行字符重建,并通过 ML 估计bootstrap 树,以确定 HCV 亚型的地理起源和地理区域之间具有统计学意义的扩散途径。系统发生地理分析追踪了塞浦路斯一般人群和 PWID 中亚型的起源,这些起源来自独特和重叠的全球分布区域。核心-E1 的系统发育分析表明,塞浦路斯被监禁人群的大多数序列与一般人群和 PWID 聚集在一起。我们估计,塞浦路斯的 HCV 感染源自多个全球来源,而大多数被监禁个体之间的 HCV 传播是本地发生的。这项分析是使用全球数据集追踪 HCV 传播模式的少数研究之一,这些实践和发现应该为未来的预防政策如何针对 HCV 流行提供信息。