Centre de recherche sur l'inflammation UMR 1149, Inserm - Université Paris Diderot, 75018, Paris, France.
Data Team, Département d'informatique de l'ENS, École normale supérieure, CNRS, PSL Research University, 75005, Paris, France.
Sci Rep. 2019 Jul 17;9(1):10351. doi: 10.1038/s41598-019-46649-z.
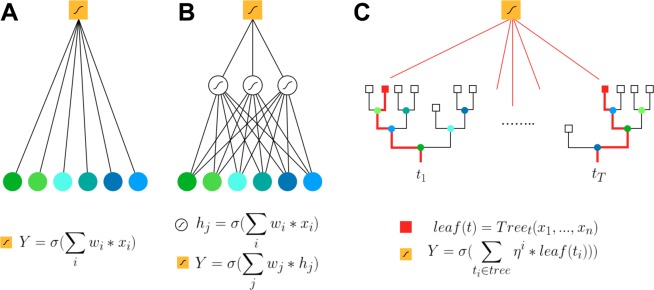
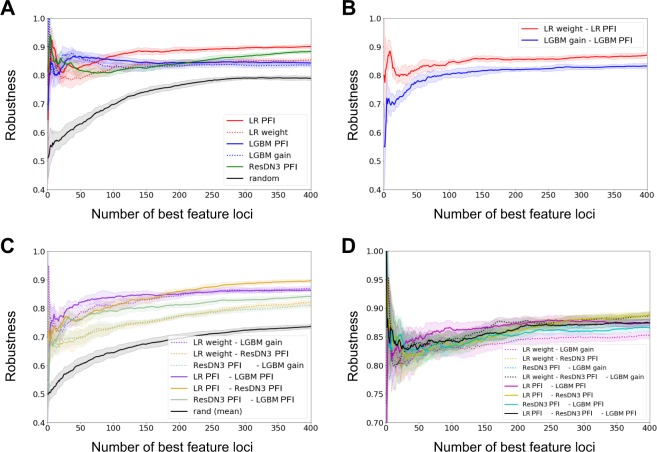
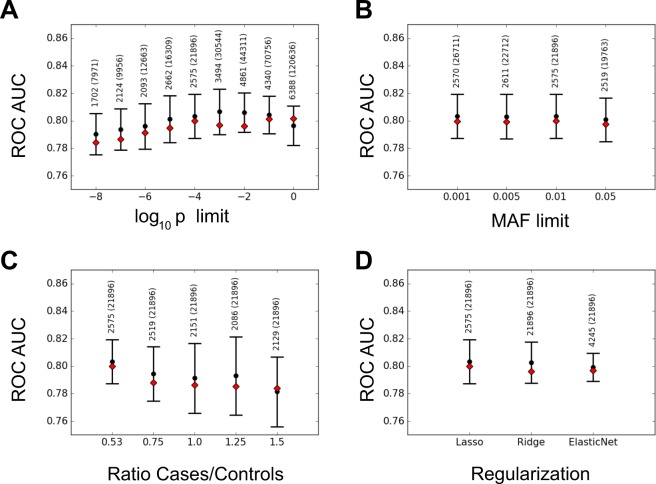
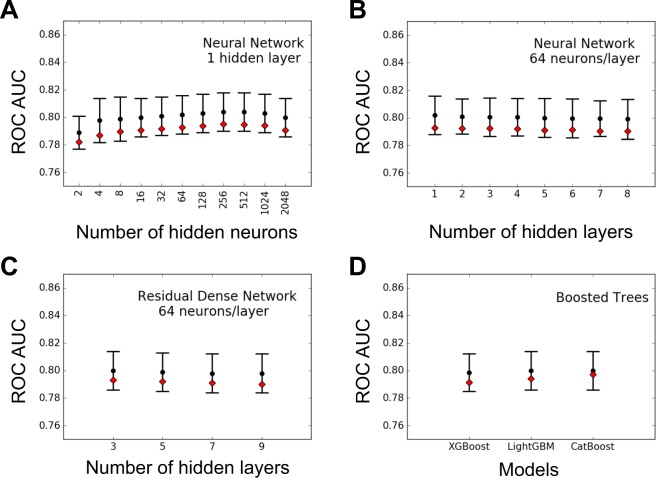
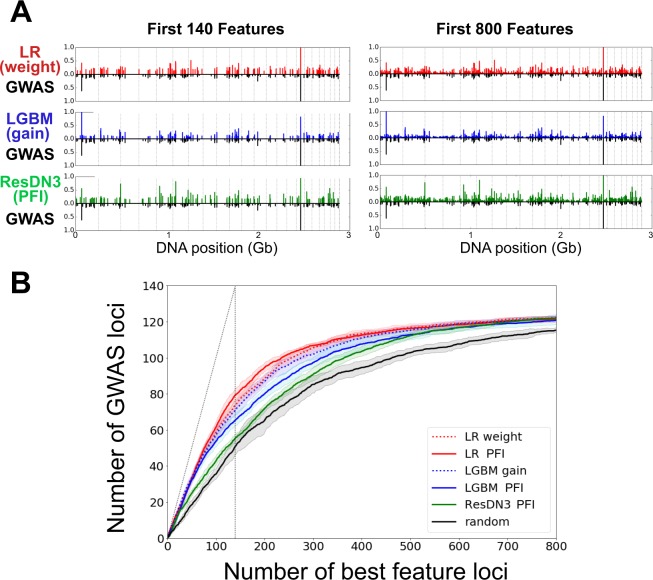
Crohn Disease (CD) is a complex genetic disorder for which more than 140 genes have been identified using genome wide association studies (GWAS). However, the genetic architecture of the trait remains largely unknown. The recent development of machine learning (ML) approaches incited us to apply them to classify healthy and diseased people according to their genomic information. The Immunochip dataset containing 18,227 CD patients and 34,050 healthy controls enrolled and genotyped by the international Inflammatory Bowel Disease genetic consortium (IIBDGC) has been re-analyzed using a set of ML methods: penalized logistic regression (LR), gradient boosted trees (GBT) and artificial neural networks (NN). The main score used to compare the methods was the Area Under the ROC Curve (AUC) statistics. The impact of quality control (QC), imputing and coding methods on LR results showed that QC methods and imputation of missing genotypes may artificially increase the scores. At the opposite, neither the patient/control ratio nor marker preselection or coding strategies significantly affected the results. LR methods, including Lasso, Ridge and ElasticNet provided similar results with a maximum AUC of 0.80. GBT methods like XGBoost, LightGBM and CatBoost, together with dense NN with one or more hidden layers, provided similar AUC values, suggesting limited epistatic effects in the genetic architecture of the trait. ML methods detected near all the genetic variants previously identified by GWAS among the best predictors plus additional predictors with lower effects. The robustness and complementarity of the different methods are also studied. Compared to LR, non-linear models such as GBT or NN may provide robust complementary approaches to identify and classify genetic markers.
克罗恩病(CD)是一种复杂的遗传疾病,通过全基因组关联研究(GWAS)已经确定了超过 140 个基因。然而,该特征的遗传结构在很大程度上仍然未知。最近机器学习(ML)方法的发展促使我们应用这些方法根据他们的基因组信息来对健康人和患者进行分类。包含 18227 名 CD 患者和 34050 名健康对照的 Immunochip 数据集,由国际炎症性肠病遗传联盟(IIBDGC)招募和基因分型,使用一组 ML 方法重新进行了分析:惩罚逻辑回归(LR)、梯度提升树(GBT)和人工神经网络(NN)。用于比较方法的主要得分是 ROC 曲线下的面积(AUC)统计数据。质量控制(QC)、缺失基因型的插补和编码方法对 LR 结果的影响表明,QC 方法和缺失基因型的插补可能会人为地增加分数。相反,患者/对照比例、标记物预选或编码策略都不会显著影响结果。LR 方法,包括 Lasso、Ridge 和 ElasticNet,提供了相似的结果,最大 AUC 为 0.80。像 XGBoost、LightGBM 和 CatBoost 这样的 GBT 方法,以及具有一个或多个隐藏层的密集 NN,提供了相似的 AUC 值,这表明在该特征的遗传结构中,上位效应有限。ML 方法检测到了先前 GWAS 确定的所有遗传变异中的近一半,以及其他具有较低效应的预测因子。还研究了不同方法的稳健性和互补性。与 LR 相比,非线性模型(如 GBT 或 NN)可能提供稳健的互补方法来识别和分类遗传标记。