Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles, California, USA.
Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Los Angeles, California, USA
mBio. 2019 Jul 23;10(4):e01660-19. doi: 10.1128/mBio.01660-19.
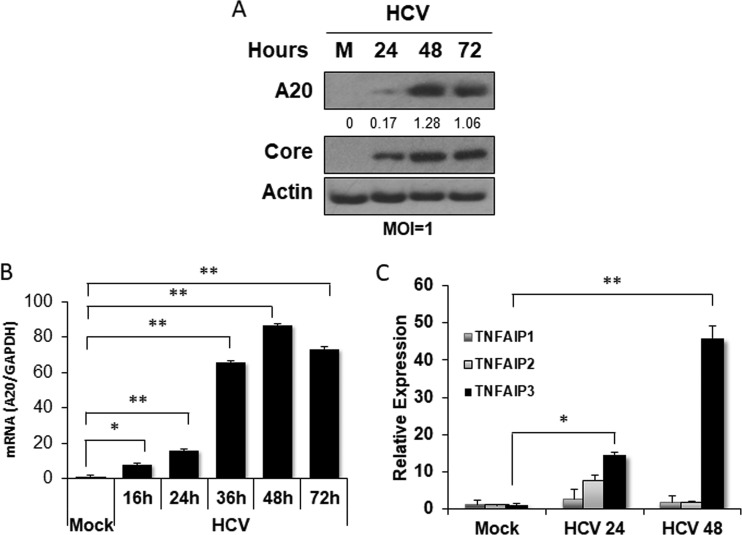
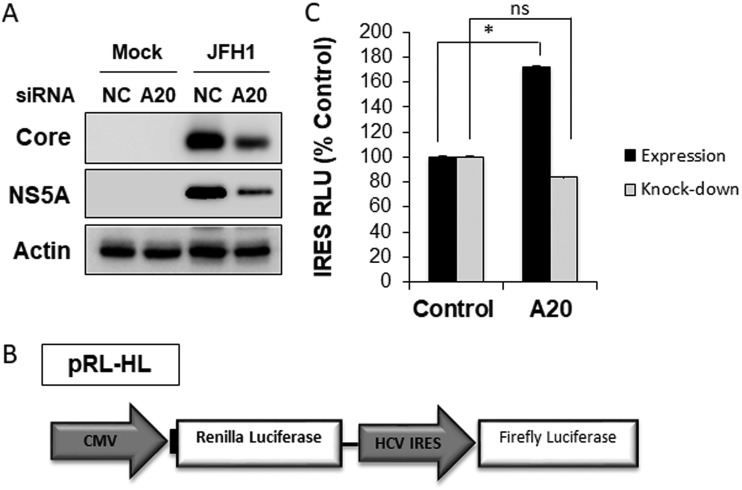
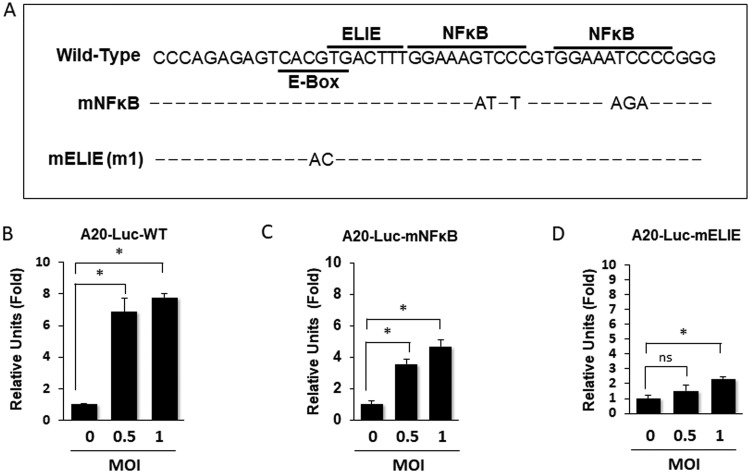
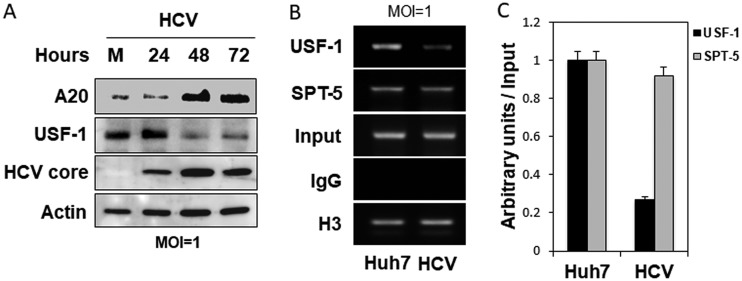
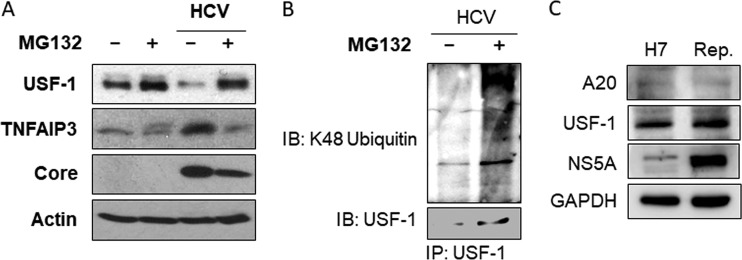
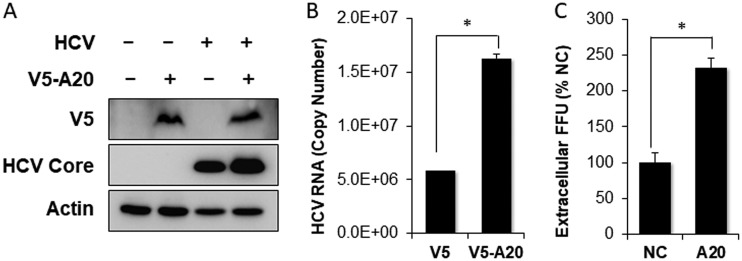
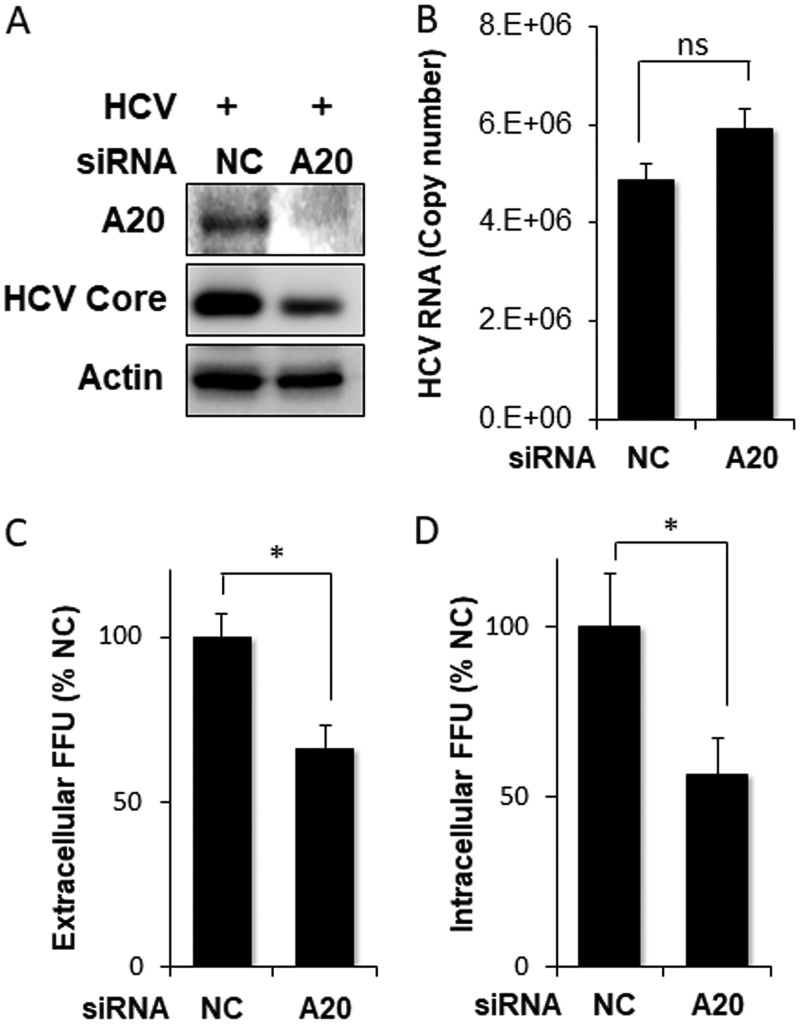
Tumor necrosis factor alpha-induced protein 3 (TNFAIP3), also known as A20, is a ubiquitin-editing enzyme capable of ubiquitination or deubiquitination of its target proteins. In this study, we show that hepatitis C virus (HCV) infection could induce the expression of A20 via the activation of the A20 promoter. The induction of A20 by HCV coincided with the loss of upstream stimulatory factor 1 (USF-1), a transcription factor known to suppress the A20 promoter. The role of USF-1 in the regulation of the A20 promoter in HCV-infected cells was confirmed by the chromatin immunoprecipitation (ChIP) assay, and its depletion was apparently mediated by proteasomes, as USF-1 could be stabilized by the proteasome inhibitor MG132 to suppress the A20 expression. As the overexpression of A20 enhanced the replication of HCV and the silencing of A20 had the opposite effect, A20 is a positive regulator of HCV replication. Our further studies indicated that A20 enhanced the activity of the HCV internal ribosome entry site (IRES). In conclusion, our results demonstrated that HCV could induce the expression of A20 via the depletion of USF-1 to enhance its replication. Our study provided important information for further understanding the interaction between HCV and its host cells. Hepatitis C virus establishes chronic infection in approximately 85% of the patients whom it infects. However, the mechanism of how HCV evades host immunity to establish persistence is unclear. In this report, we demonstrate that HCV could induce the expression of the ubiquitin-editing enzyme A20, an important negative regulator of the tumor necrosis factor alpha (TNF-α) and NF-κB signaling pathways. This induction of A20 enhanced HCV replication as it could stimulate the HCV IRES activity to enhance the translation of HCV proteins. The induction of A20 was mediated by the depletion of USF-1, a suppressor of the A20 promoter. Our study thus provides important information for further understanding the interaction between HCV and its host cells.
肿瘤坏死因子-α诱导蛋白 3(TNFAIP3),也称为 A20,是一种具有泛素化或去泛素化其靶蛋白能力的泛素编辑酶。在这项研究中,我们表明 HCV 感染可以通过激活 A20 启动子诱导 A20 的表达。HCV 诱导的 A20 与上游刺激因子 1(USF-1)的丧失同时发生,USF-1 是一种已知抑制 A20 启动子的转录因子。通过染色质免疫沉淀(ChIP)实验证实了 USF-1 在 HCV 感染细胞中调节 A20 启动子的作用,并且其耗竭显然是通过蛋白酶体介导的,因为 USF-1 可以被蛋白酶体抑制剂 MG132 稳定以抑制 A20 的表达。由于 A20 的过表达增强了 HCV 的复制,而 A20 的沉默则产生相反的效果,因此 A20 是 HCV 复制的正调节剂。我们的进一步研究表明,A20 增强了 HCV 内部核糖体进入位点(IRES)的活性。总之,我们的结果表明,HCV 通过耗尽 USF-1 诱导 A20 的表达来增强其复制。我们的研究为进一步了解 HCV 与其宿主细胞之间的相互作用提供了重要信息。HCV 在其感染的大约 85%的患者中建立慢性感染。然而,HCV 如何逃避宿主免疫以建立持续性的机制尚不清楚。在本报告中,我们证明 HCV 可以诱导泛素编辑酶 A20 的表达,A20 是肿瘤坏死因子-α(TNF-α)和 NF-κB 信号通路的重要负调节剂。这种 A20 的诱导增强了 HCV 的复制,因为它可以刺激 HCV IRES 活性以增强 HCV 蛋白的翻译。A20 的诱导是通过 USF-1 的耗竭介导的,USF-1 是 A20 启动子的抑制剂。因此,我们的研究为进一步了解 HCV 与其宿主细胞之间的相互作用提供了重要信息。