Chandra Goutam, Defour Aurelia, Mamchoui Kamel, Pandey Kalpana, Mishra Soumya, Mouly Vincent, Sreetama SenChandra, Mahad Ahmad Mohammad, Mahjneh Ibrahim, Morizono Hiroki, Pattabiraman Nagarajan, Menon Anant K, Jaiswal Jyoti K
1Center of Genetic Medicine Research, Children's National Health System, 111 Michigan Avenue, NW, Washington, DC 20010 USA.
7Present Address: Aix Marseille Université, UMR_S 910, Génétique Médicale et Génomique Fonctionnelle, 13385 Marseille, France.
Cell Death Discov. 2019 Jul 18;5:118. doi: 10.1038/s41420-019-0197-z. eCollection 2019.
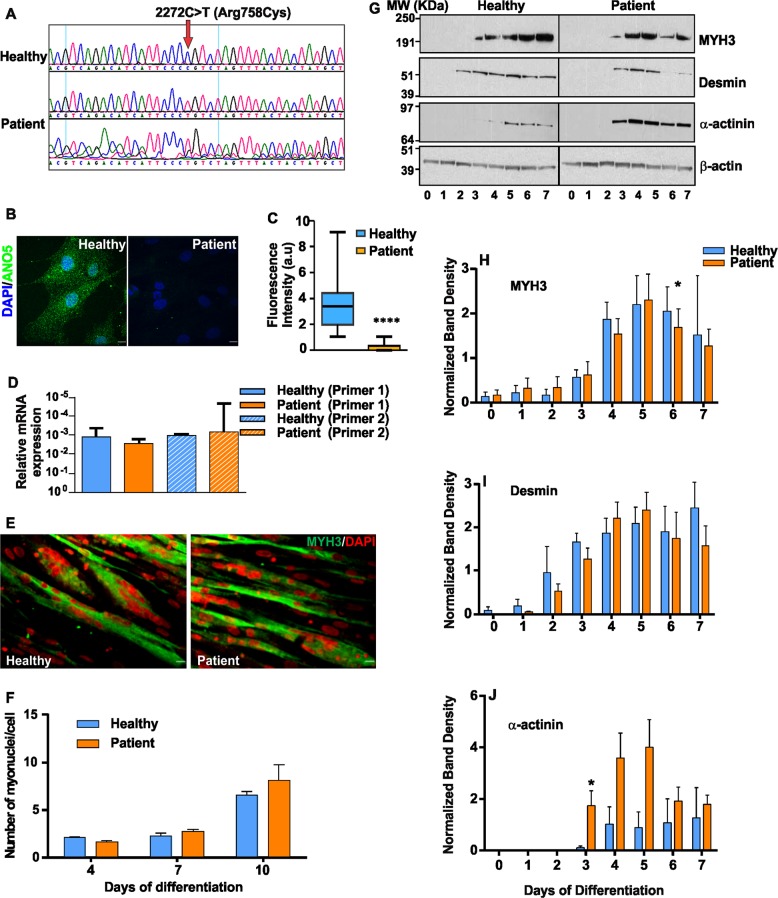
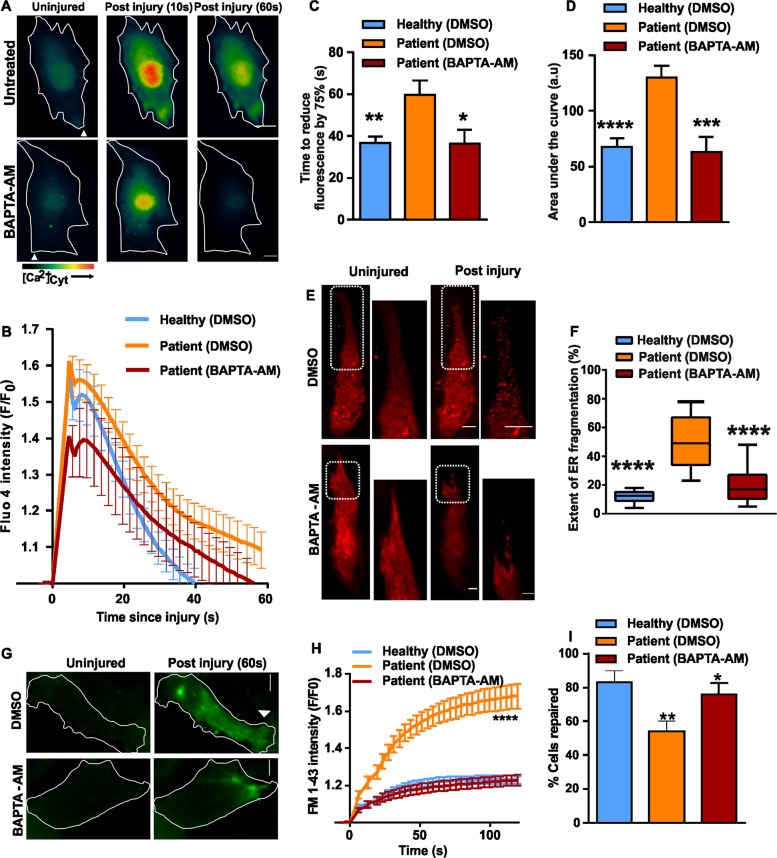
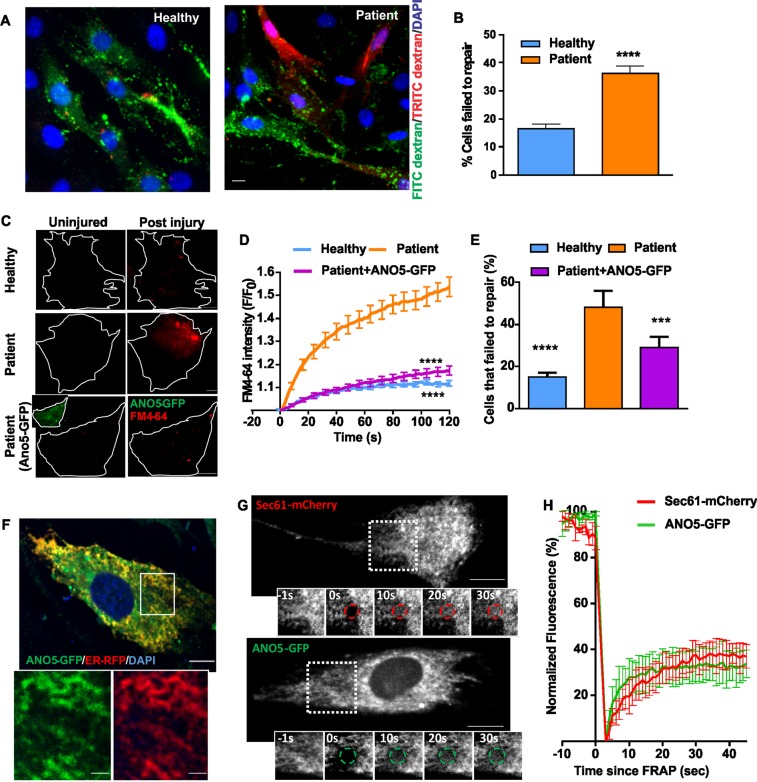
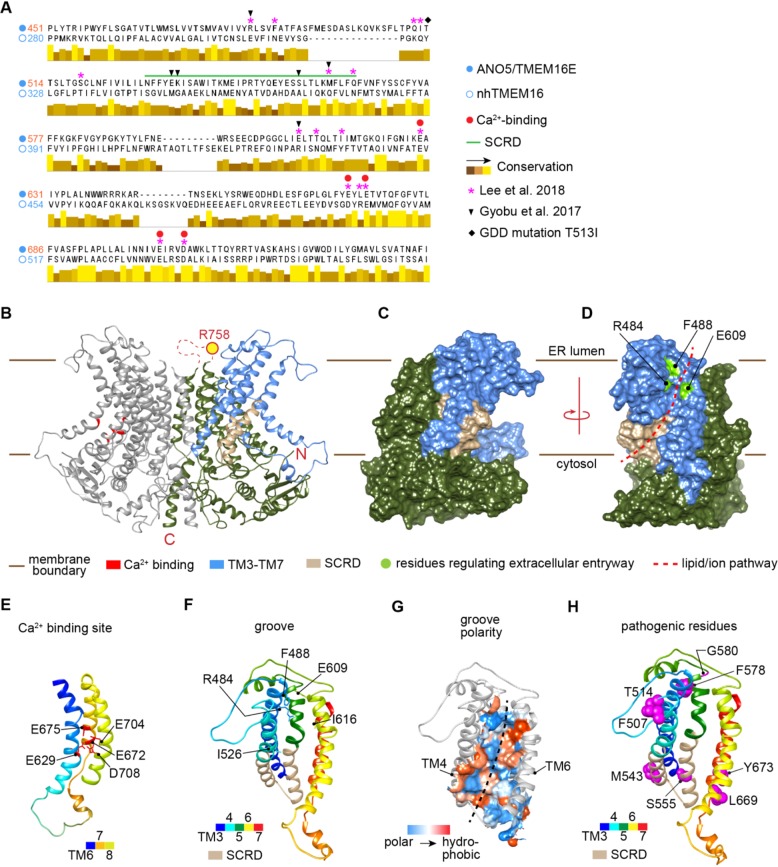
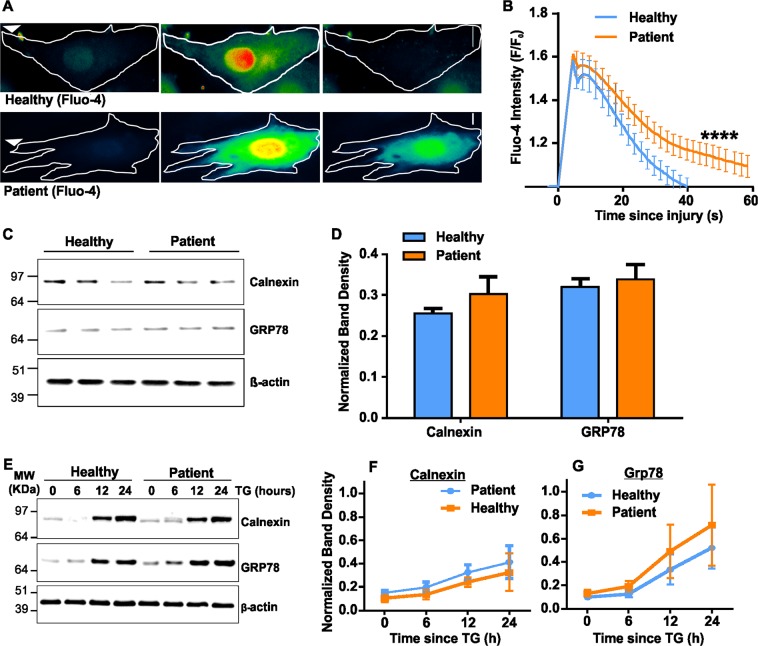
Autosomal recessive mutations in Anoctamin 5 (), a member of the transmembrane 16 (TMEM16) family of Ca-activated ion channels and phospholipid scramblases, cause adult-onset muscular dystrophies (limb girdle muscular dystrophy 2L (LGMD2L) and Miyoshi Muscular Dystrophy (MMD3). However, the molecular role of ANO5 is unclear and knockout mouse models show conflicting requirements of ANO5 in muscle. To study the role of ANO5 in human muscle cells we generated a myoblast line from a MMD3-patient carrying the c.2272C>T mutation, which we find causes the mutant protein to be degraded. The patient myoblasts exhibit normal myogenesis, but are compromised in their plasma membrane repair (PMR) ability. The repair deficit is linked to the poor ability of the endoplasmic reticulum (ER) to clear cytosolic Ca increase caused by focal plasma membrane injury. Expression of wild-type ANO5 or pharmacological prevention of injury-triggered cytosolic Ca overload enable injured patient muscle cells to repair. A homology model of ANO5 shows that several of the known LGMD2L/MMD3 patient mutations line the transmembrane region of the protein implicated in its channel activity. These results point to a role of cytosolic Ca homeostasis in PMR, indicate a role for ANO5 in ER-mediated cytosolic Ca uptake and identify normalization of cytosolic Ca homeostasis as a potential therapeutic approach to treat muscular dystrophies caused by ANO5 deficit.
anoctamin 5(ANO5)是跨膜16(TMEM16)家族中钙激活离子通道和磷脂翻转酶的成员,常染色体隐性突变会导致成人期肌营养不良(肢带型肌营养不良2L型(LGMD2L)和宫下型肌营养不良(MMD3))。然而,ANO5的分子作用尚不清楚,基因敲除小鼠模型在肌肉中对ANO5的需求也存在矛盾。为了研究ANO5在人类肌肉细胞中的作用,我们从一名携带c.2272C>T突变的MMD3患者身上生成了一个成肌细胞系,我们发现该突变导致突变蛋白被降解。患者的成肌细胞表现出正常的肌生成,但质膜修复(PMR)能力受损。修复缺陷与内质网(ER)清除局部质膜损伤引起的胞质钙增加的能力较差有关。野生型ANO5的表达或对损伤引发的胞质钙超载的药物预防可使受损的患者肌肉细胞得以修复。ANO5的同源模型表明,几个已知的LGMD2L/MMD3患者突变位于与其通道活性相关的蛋白质跨膜区域。这些结果表明胞质钙稳态在PMR中的作用,表明ANO5在内质网介导的胞质钙摄取中的作用,并确定将胞质钙稳态正常化作为治疗由ANO5缺陷引起的肌营养不良的潜在治疗方法。