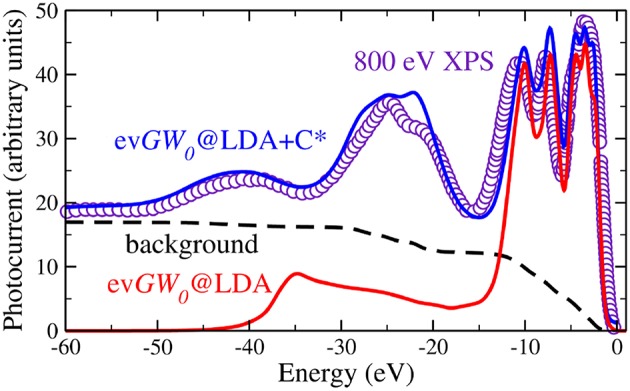
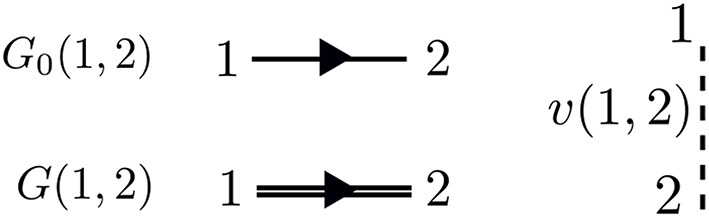
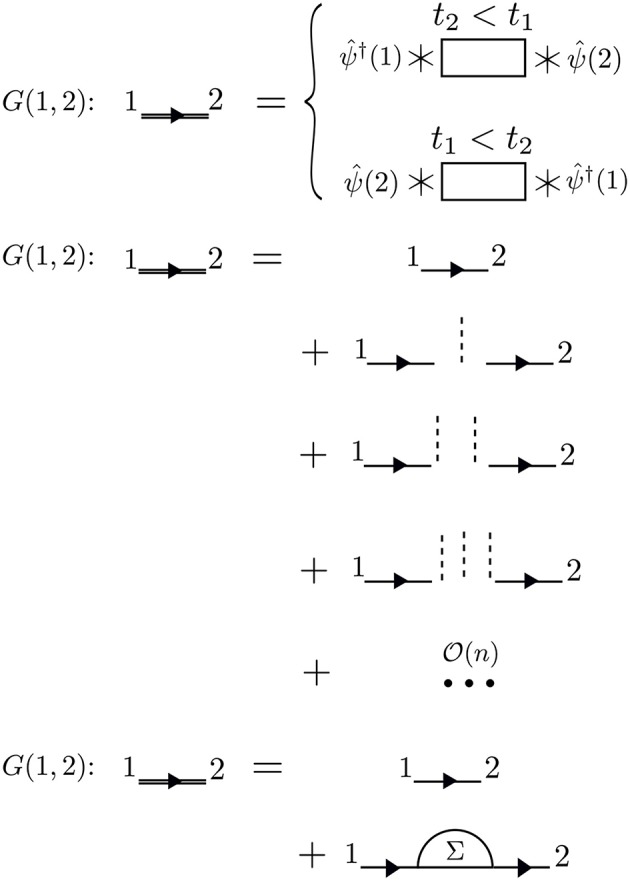
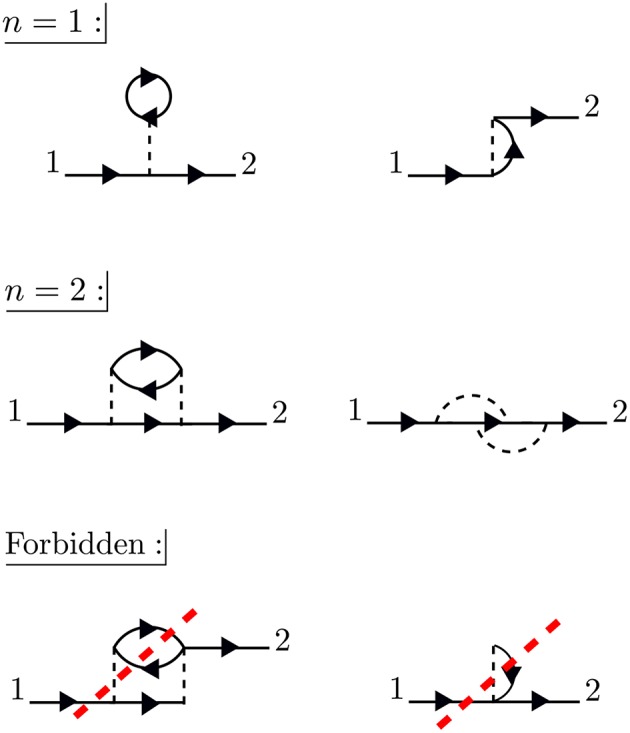
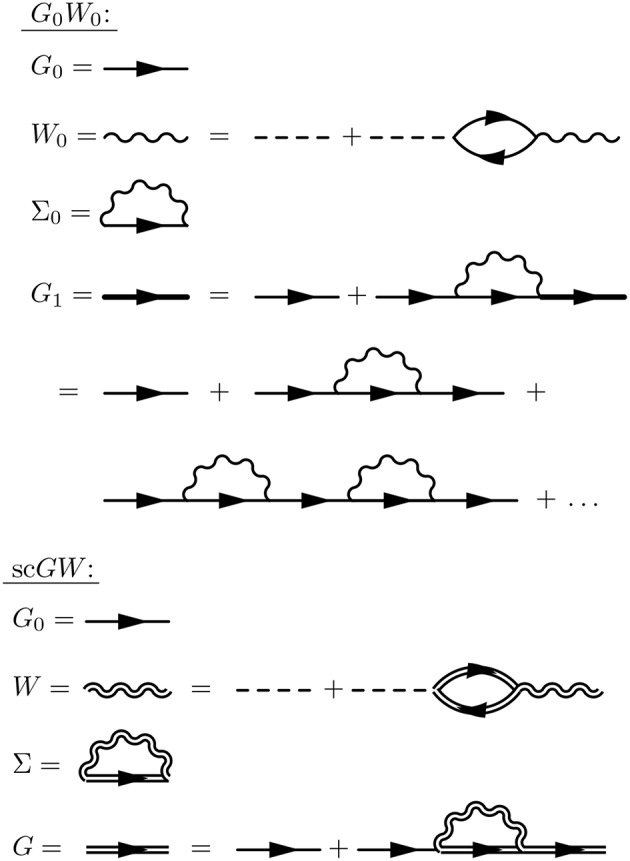
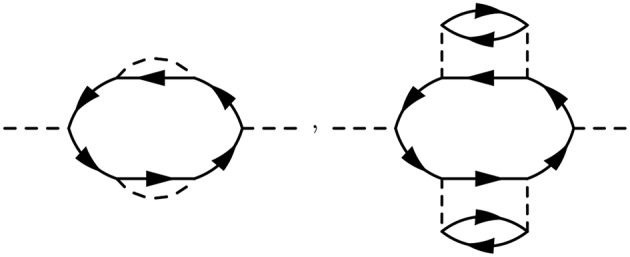
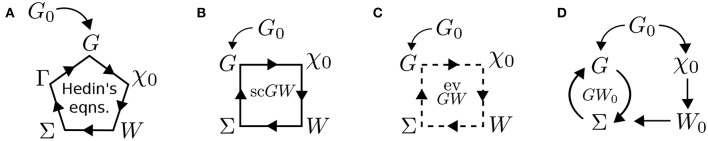
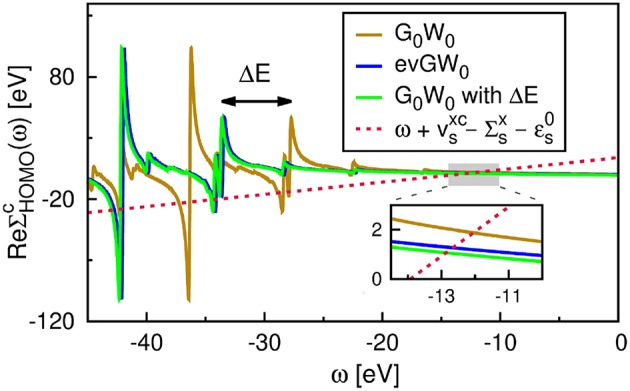
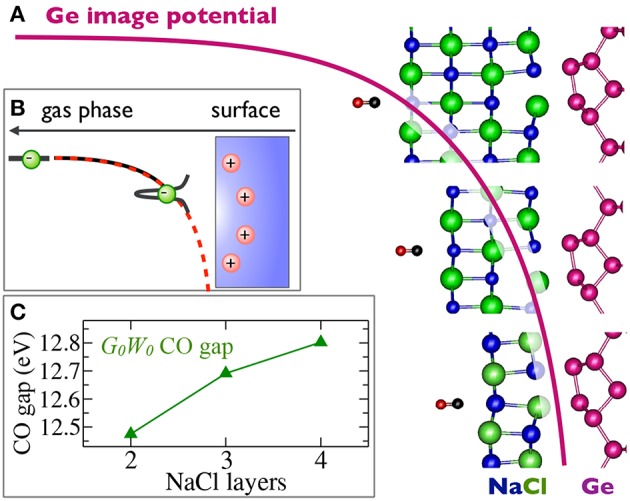
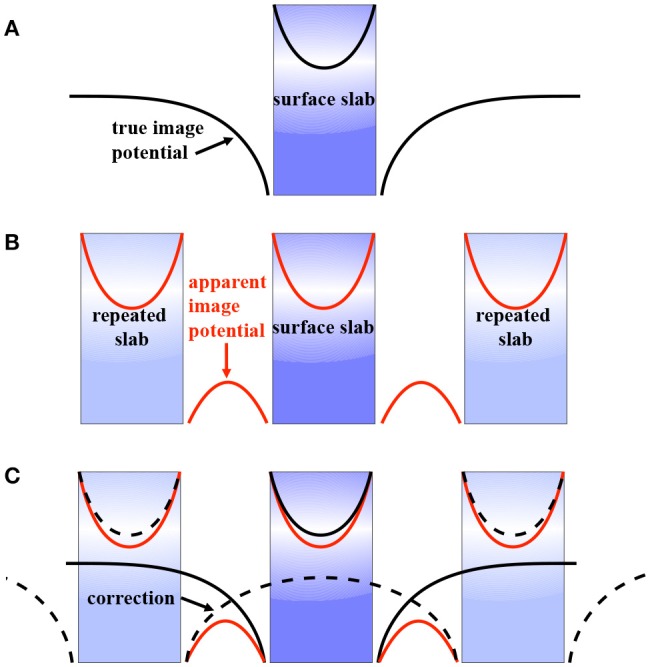
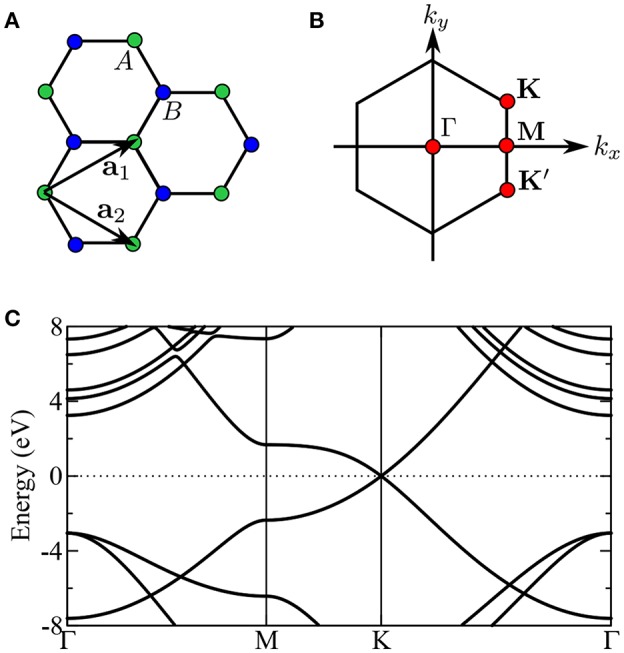
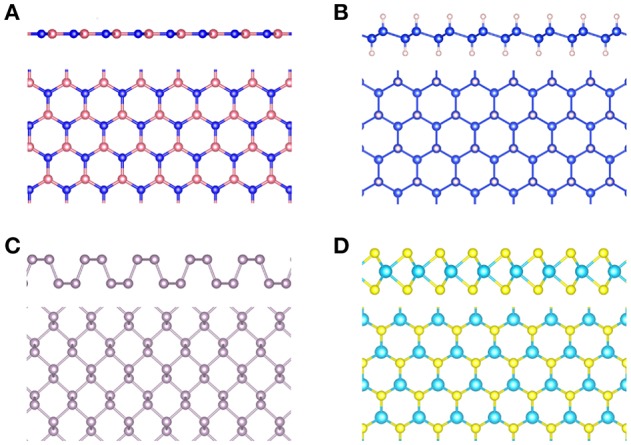
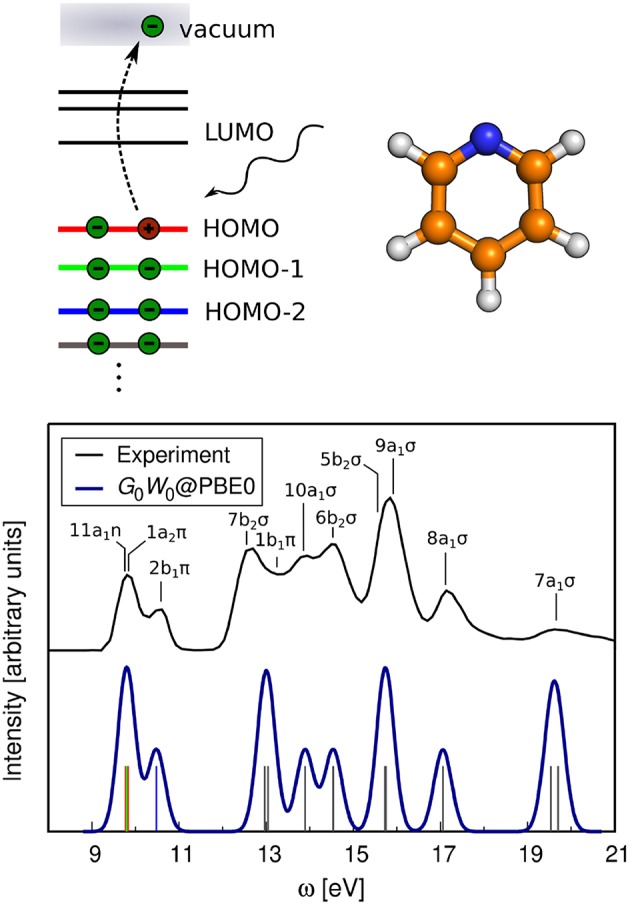
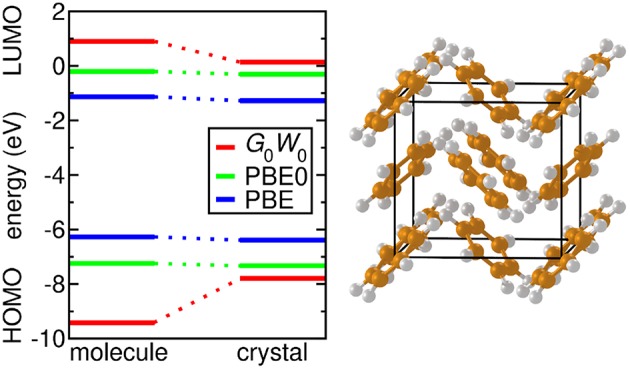
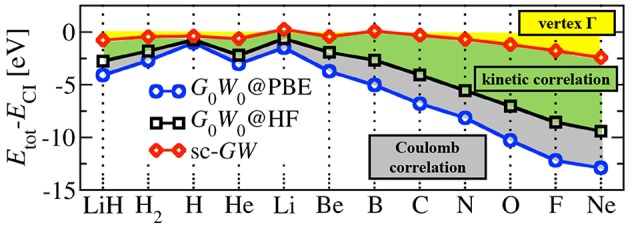
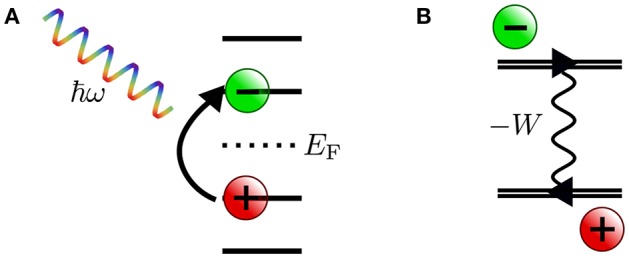
Golze Dorothea, Dvorak Marc, Rinke Patrick
Department of Applied Physics, Aalto University, School of Science, Espoo, Finland.
Front Chem. 2019 Jul 9;7:377. doi: 10.3389/fchem.2019.00377. eCollection 2019.
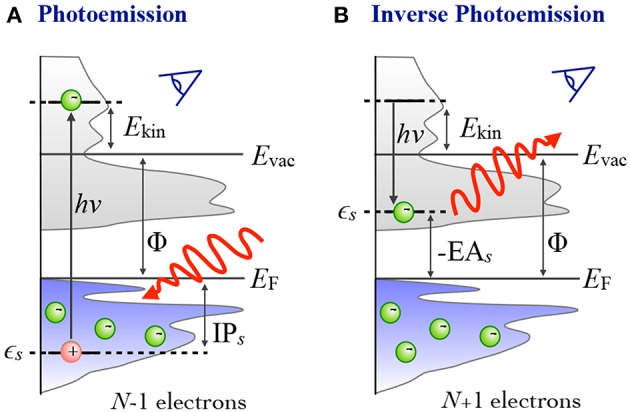
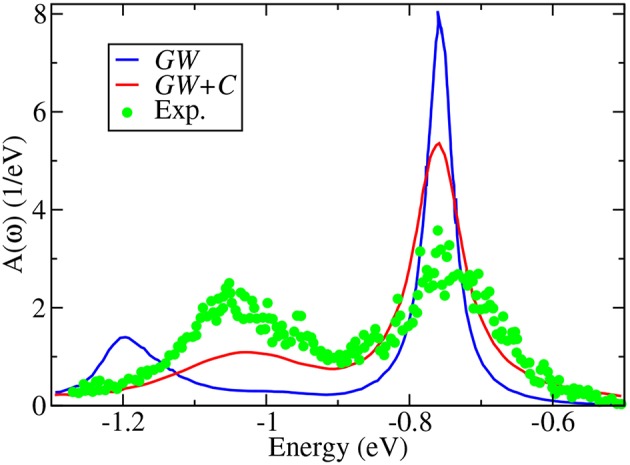
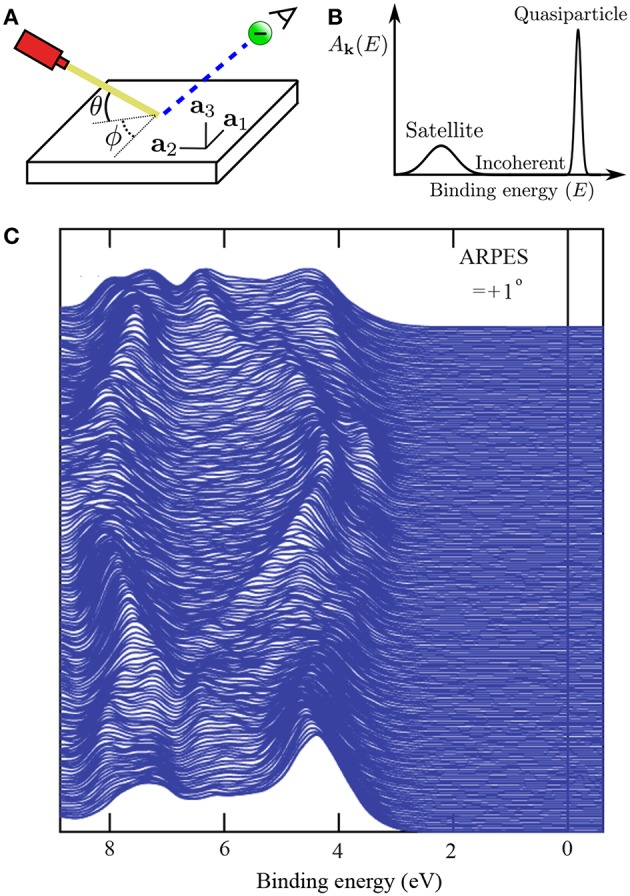
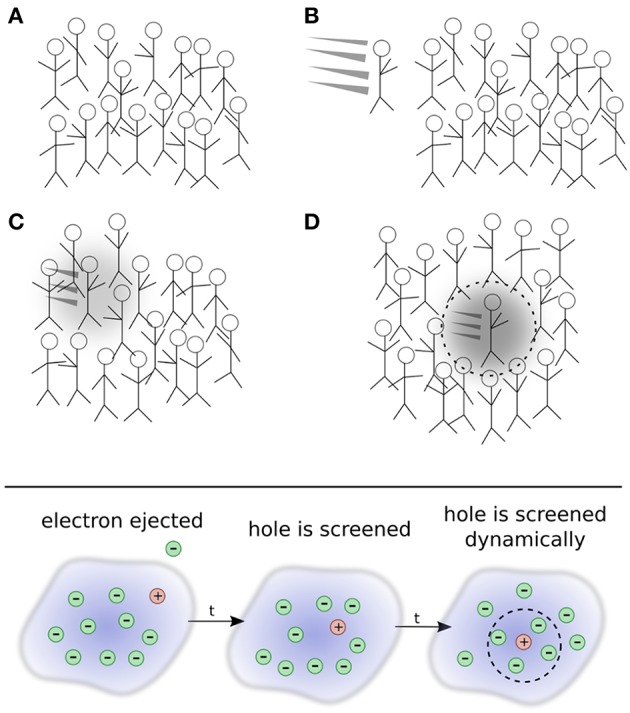
The approximation in electronic structure theory has become a widespread tool for predicting electronic excitations in chemical compounds and materials. In the realm of theoretical spectroscopy, the method provides access to charged excitations as measured in direct or inverse photoemission spectroscopy. The number of calculations in the past two decades has exploded with increased computing power and modern codes. The success of can be attributed to many factors: favorable scaling with respect to system size, a formal interpretation for charged excitation energies, the importance of dynamical screening in real systems, and its practical combination with other theories. In this review, we provide an overview of these formal and practical considerations. We expand, in detail, on the choices presented to the scientist performing calculations for the first time. We also give an introduction to the many-body theory behind , a review of modern applications like molecules and surfaces, and a perspective on methods which go beyond conventional calculations. This review addresses chemists, physicists and material scientists with an interest in theoretical spectroscopy. It is intended for newcomers to calculations but can also serve as an alternative perspective for experts and an up-to-date source of computational techniques.
电子结构理论中的这种近似方法已成为预测化合物和材料中电子激发的广泛应用工具。在理论光谱学领域,该方法可用于获取直接或逆光电子能谱中所测量的带电激发。在过去二十年中,随着计算能力的提升和现代程序的发展,此类计算的数量激增。该方法的成功可归因于诸多因素:在系统尺寸方面具有良好的标度性、对带电激发能有形式化解释、实际系统中动态屏蔽的重要性以及它与其他理论的实际结合。在本综述中,我们概述了这些形式化和实际的考量因素。我们详细阐述了首次进行此类计算的科学家所面临的选择。我们还介绍了该方法背后的多体理论,回顾了分子和表面等现代应用,并展望了超越传统此类计算的方法。本综述面向对理论光谱学感兴趣的化学家、物理学家和材料科学家。它旨在为初次接触此类计算的人员提供帮助,也可为专家提供另一种视角,并作为计算技术的最新来源。