Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Oxford, UK.
School of Life and Medical Sciences, University of Hertfordshire, Hatfield, UK.
J Gen Physiol. 2019 Sep 2;151(9):1094-1115. doi: 10.1085/jgp.201912351. Epub 2019 Jul 29.
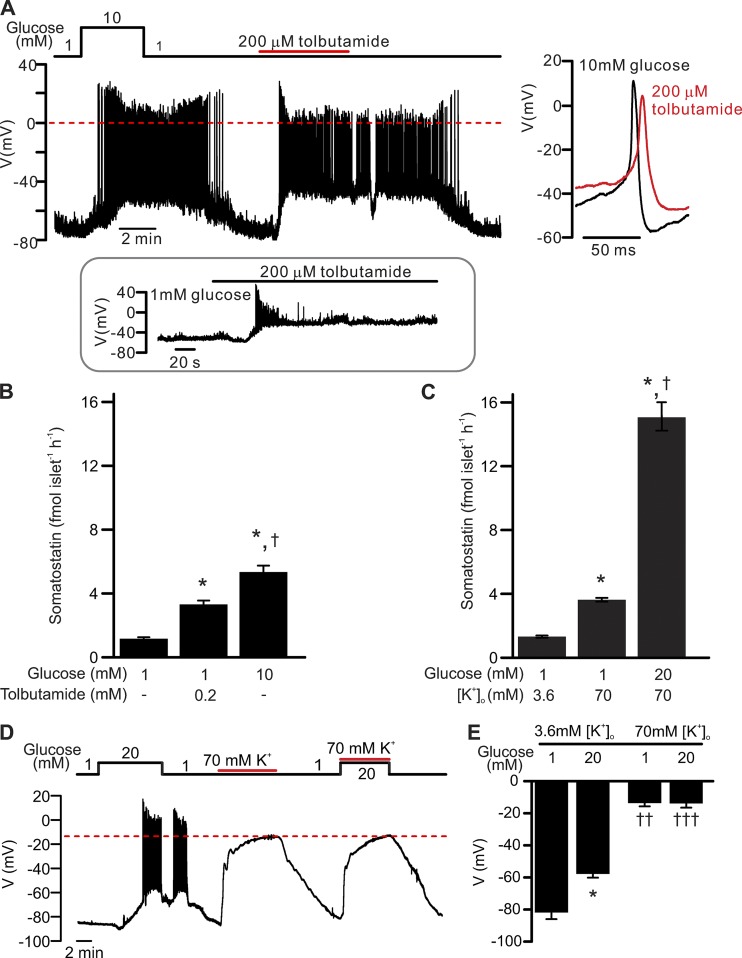
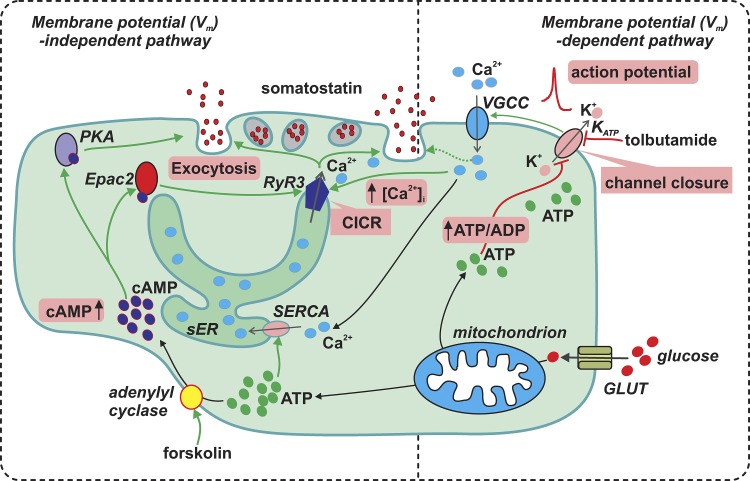
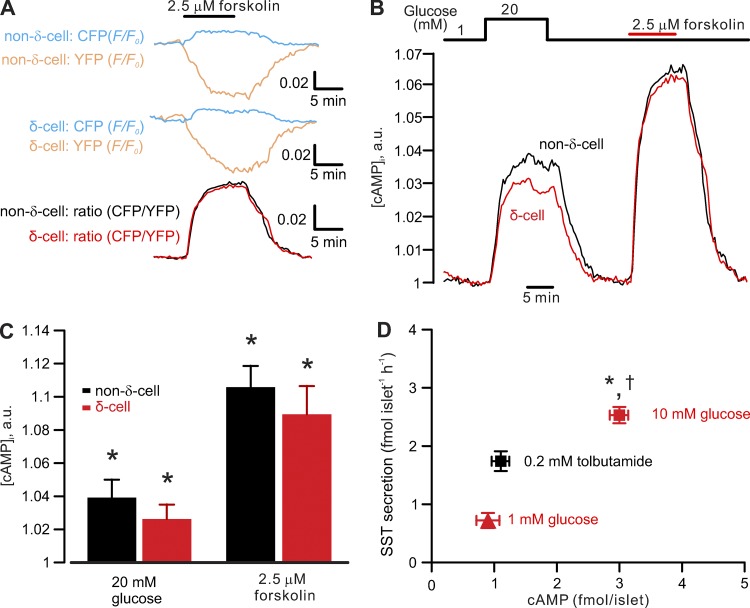
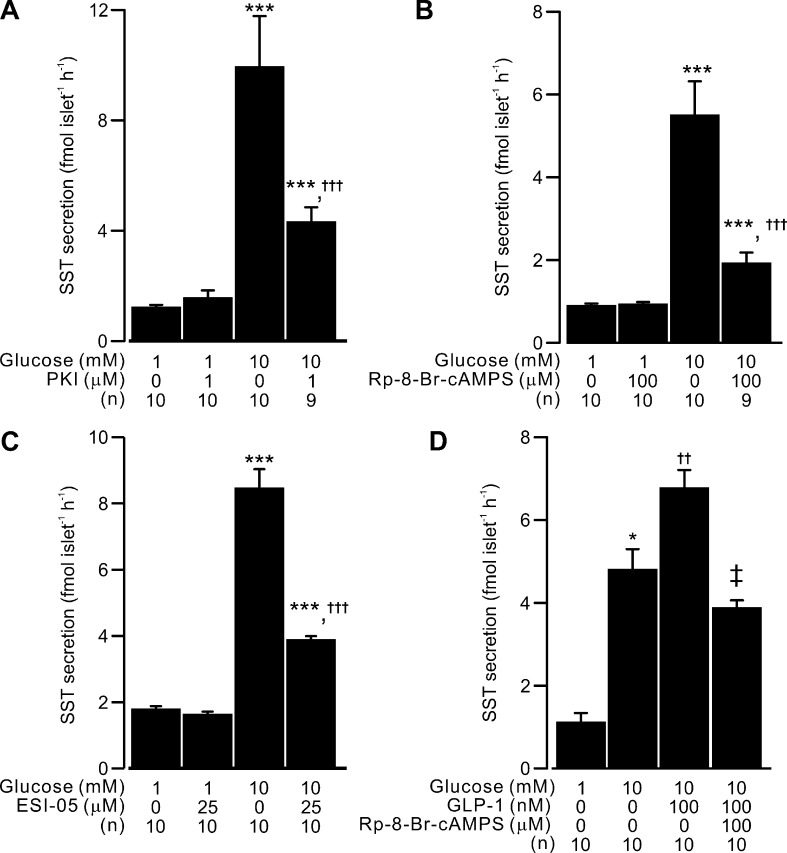
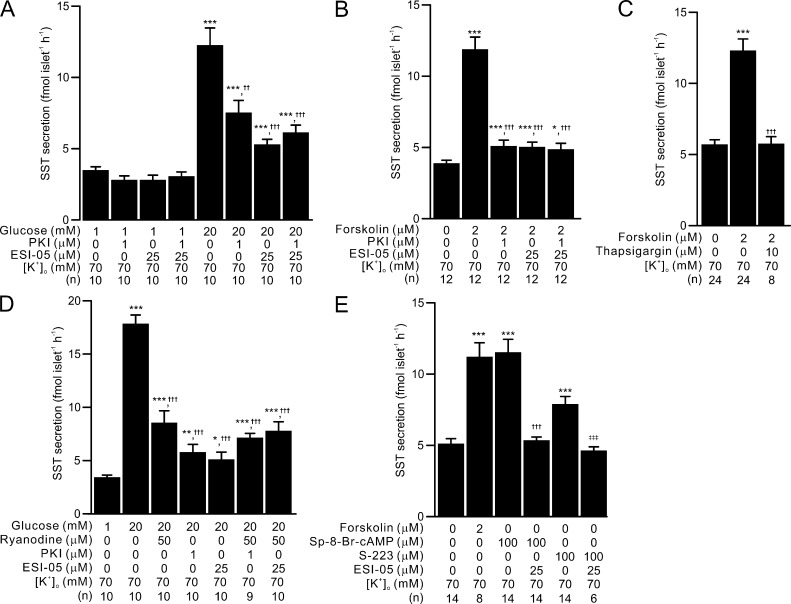
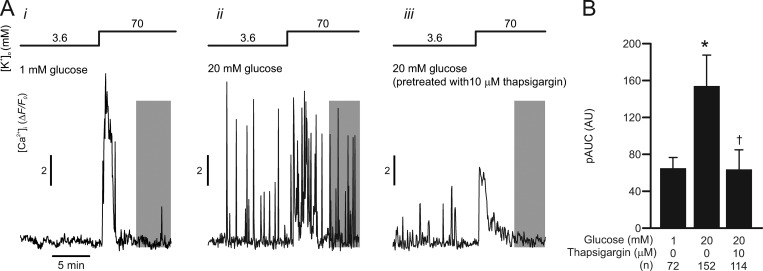
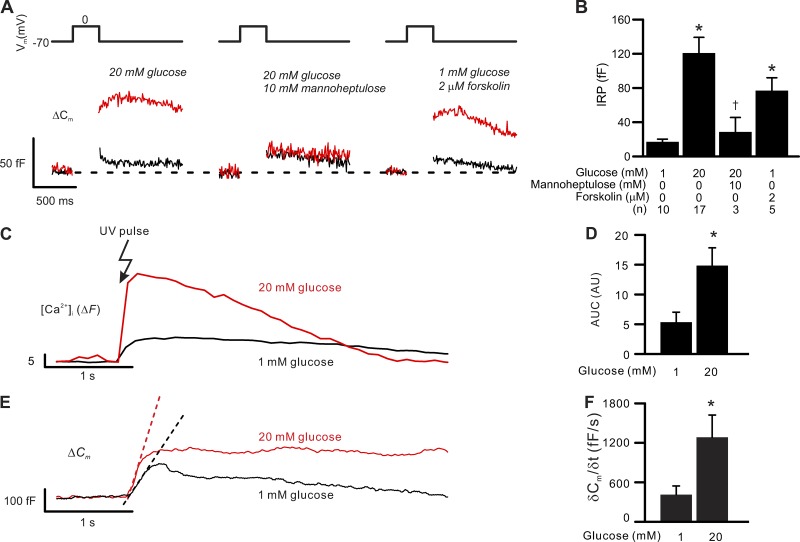
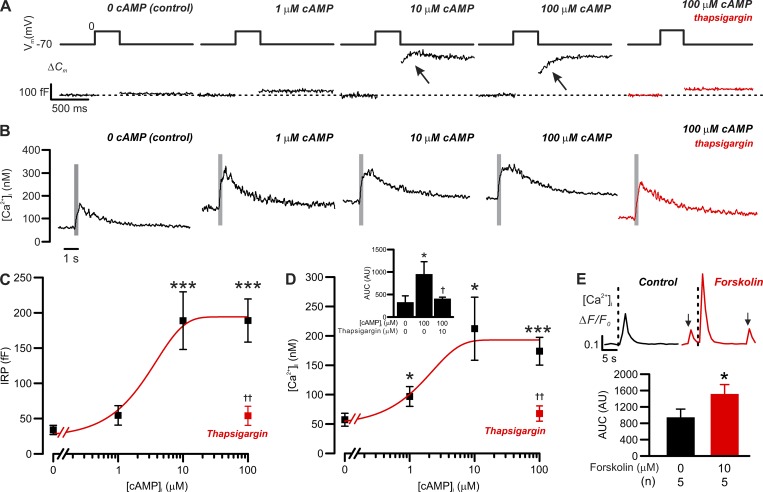
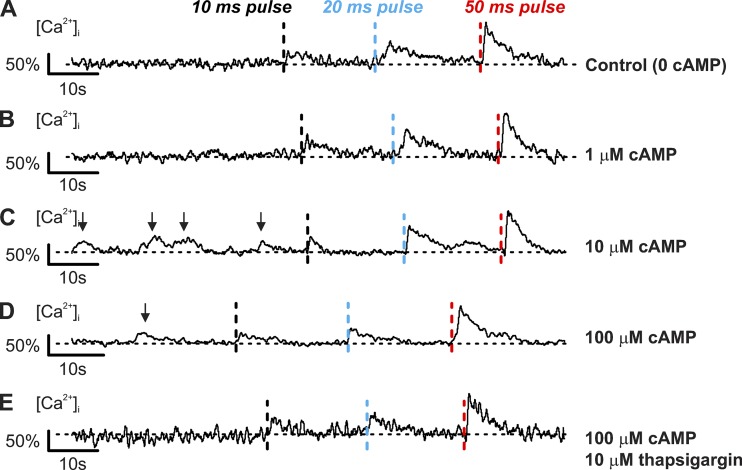
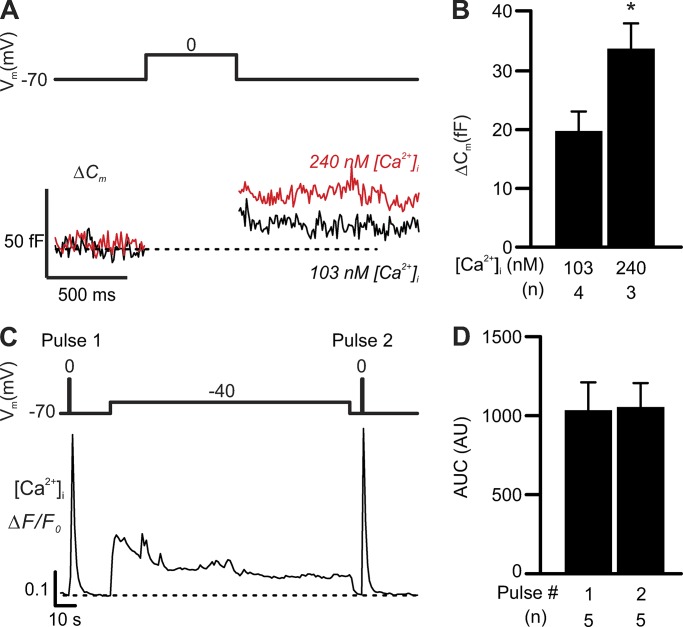
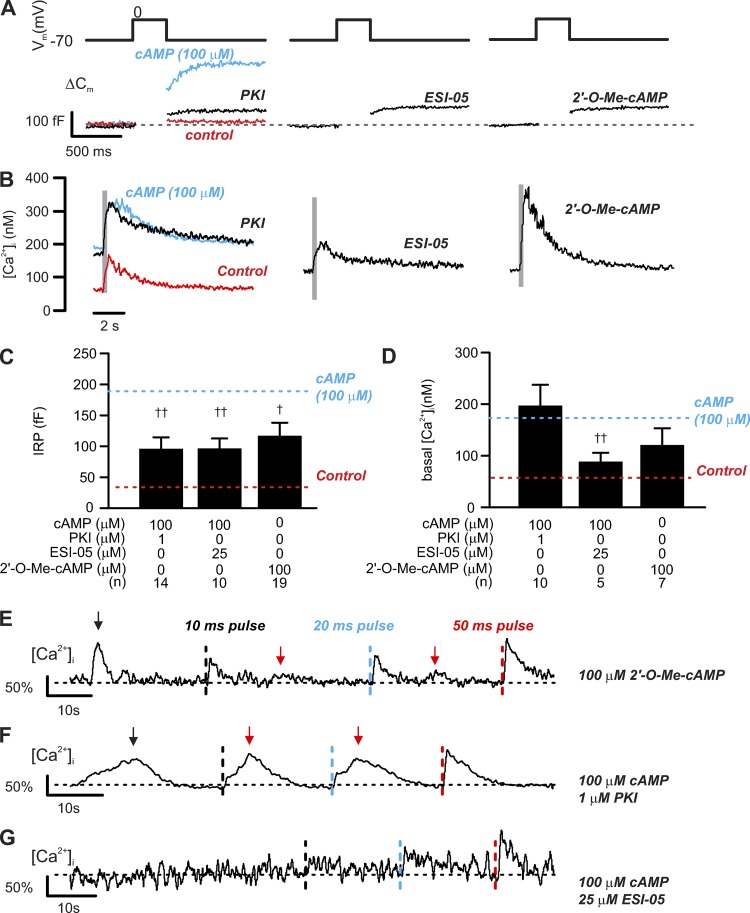
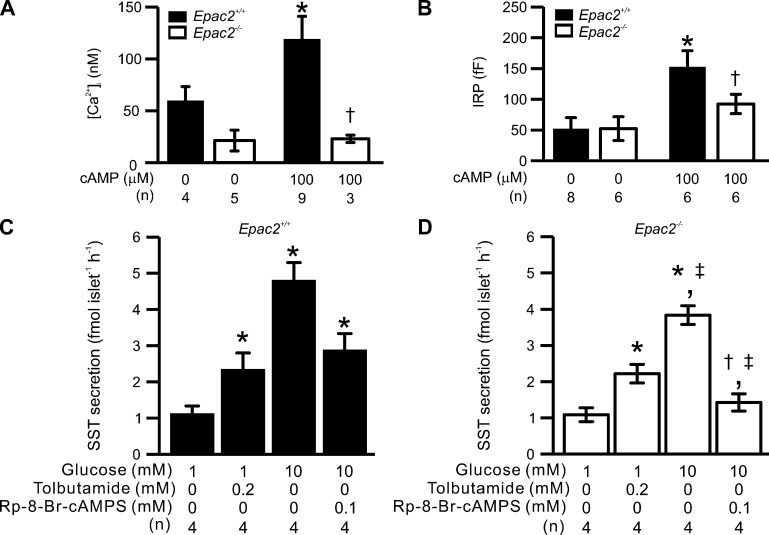
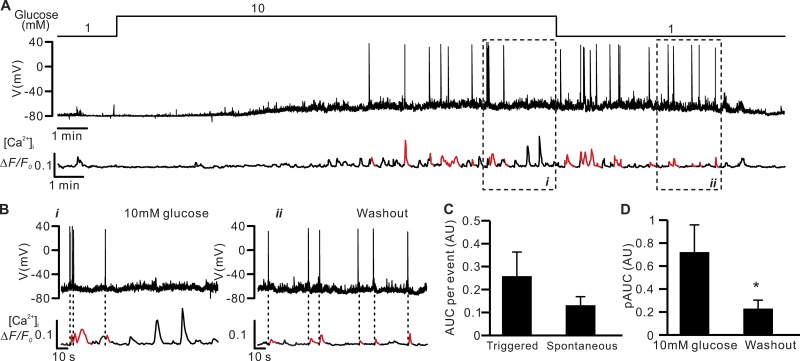
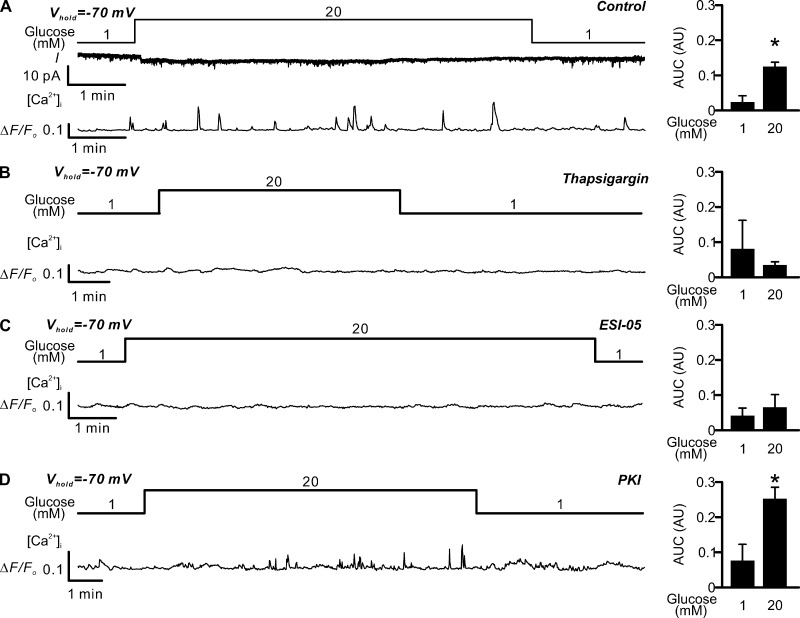
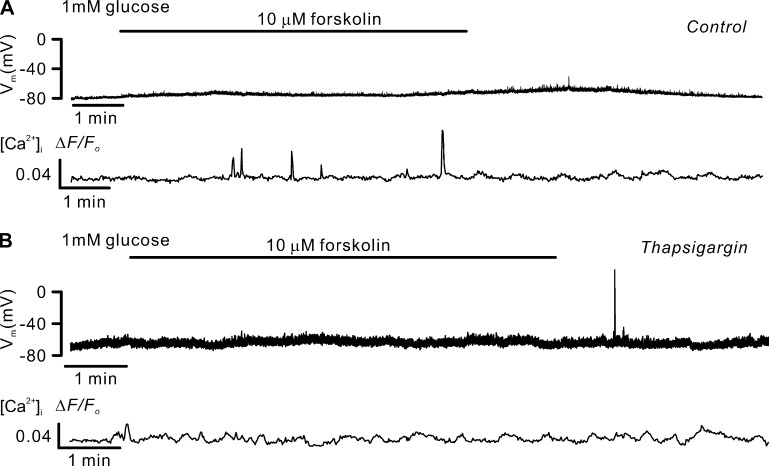
Somatostatin secretion from pancreatic islet δ-cells is stimulated by elevated glucose levels, but the underlying mechanisms have only partially been elucidated. Here we show that glucose-induced somatostatin secretion (GISS) involves both membrane potential-dependent and -independent pathways. Although glucose-induced electrical activity triggers somatostatin release, the sugar also stimulates GISS via a cAMP-dependent stimulation of CICR and exocytosis of somatostatin. The latter effect is more quantitatively important and in mouse islets depolarized by 70 mM extracellular K increasing glucose from 1 mM to 20 mM produced an ∼3.5-fold stimulation of somatostatin secretion, an effect that was mimicked by the application of the adenylyl cyclase activator forskolin. Inhibiting cAMP-dependent pathways with PKI or ESI-05, which inhibit PKA and exchange protein directly activated by cAMP 2 (Epac2), respectively, reduced glucose/forskolin-induced somatostatin secretion. Ryanodine produced a similar effect that was not additive to that of the PKA or Epac2 inhibitors. Intracellular application of cAMP produced a concentration-dependent stimulation of somatostatin exocytosis and elevation of cytoplasmic Ca ([Ca]). Both effects were inhibited by ESI-05 and thapsigargin (an inhibitor of SERCA). By contrast, inhibition of PKA suppressed δ-cell exocytosis without affecting [Ca] Simultaneous recordings of electrical activity and [Ca] in δ-cells expressing the genetically encoded Ca indicator GCaMP3 revealed that the majority of glucose-induced [Ca] spikes did not correlate with δ-cell electrical activity but instead reflected Ca release from the ER. These spontaneous [Ca] spikes are resistant to PKI but sensitive to ESI-05 or thapsigargin. We propose that cAMP links an increase in plasma glucose to stimulation of somatostatin secretion by promoting CICR, thus evoking exocytosis of somatostatin-containing secretory vesicles in the δ-cell.
胰岛 δ 细胞的生长抑素分泌受高血糖水平刺激,但潜在机制尚未完全阐明。本研究表明,葡萄糖诱导的生长抑素分泌(GISS)涉及膜电位依赖和非依赖途径。尽管葡萄糖诱导的电活动触发生长抑素释放,但该糖还通过 cAMP 依赖性刺激钙释放和生长抑素的胞吐作用刺激 GISS。后一种效应更为重要,在小鼠胰岛中,70mM 的细胞外 K+ 去极化将葡萄糖从 1mM 增加到 20mM 可使生长抑素分泌增加约 3.5 倍,这一效应可被腺苷酸环化酶激活剂 forskolin 模拟。用 PKI 或 ESI-05 抑制 cAMP 依赖性途径,分别抑制 PKA 和 cAMP 直接激活的交换蛋白 2(Epac2),可降低葡萄糖/ forskolin 诱导的生长抑素分泌。Ryanodine 产生了类似的效应,但与 PKA 或 Epac2 抑制剂的效应无叠加。细胞内应用 cAMP 可浓度依赖性地刺激生长抑素胞吐作用并升高细胞质 Ca([Ca])。这两种效应均被 ESI-05 和 thapsigargin(SERCA 抑制剂)抑制。相比之下,抑制 PKA 抑制 δ 细胞胞吐作用而不影响 [Ca]。在表达基因编码 Ca 指示剂 GCaMP3 的 δ 细胞中同时记录电活动和 [Ca],发现大多数葡萄糖诱导的 [Ca] 峰与 δ 细胞电活动无关,而是反映内质网 Ca 释放。这些自发的 [Ca] 峰对 PKI 有抗性,但对 ESI-05 或 thapsigargin 敏感。我们提出 cAMP 通过促进钙释放,将血浆葡萄糖的增加与生长抑素分泌的刺激联系起来,从而在 δ 细胞中引发含生长抑素的分泌囊泡的胞吐作用。