Sahu Divya, Ho Shinn-Ying, Juan Hsueh-Fen, Huang Hsuan-Cheng
Institute of Bioinformatics and Systems Biology.
Bioinformatics Program, Taiwan International Graduate Program, Institute of Information Science, Academia Sinica, Taipei, Taiwan.
JNCI Cancer Spectr. 2018 Jun 1;2(2):pky015. doi: 10.1093/jncics/pky015. eCollection 2018 Apr.
Current clinical risk factors stratify patients with neuroblastoma (NB) for appropriate treatments, yet patients with similar clinical behaviors evoke variable responses. MYCN amplification is one of the established drivers of NB and, when combined with high-risk displays, worsens outcomes. Growing high-throughput transcriptomics studies suggest long noncoding RNA (lncRNA) dysregulation in cancers, including NB. However, expression-based lncRNA signatures are altered by MYCN amplification, which is associated with high-risk, and patient prognosis remains limited.
We investigated RNA-seq-based expression profiles of lncRNAs in MYCN status and risk status in a discovery cohort (n = 493) and validated them in three independent cohorts. In the discovery cohort, a prognostic association of lncRNAs was determined by univariate Cox regression and integrated into a signature using the risk score method. A novel risk score threshold selection criterion was developed to stratify patients into risk groups. Outcomes by risk group and clinical subgroup were assessed using Kaplan-Meier survival curves and multivariable Cox regression. The performance of lncRNA signatures was evaluated by receiver operating characteristic curve. All statistical tests were two-sided.
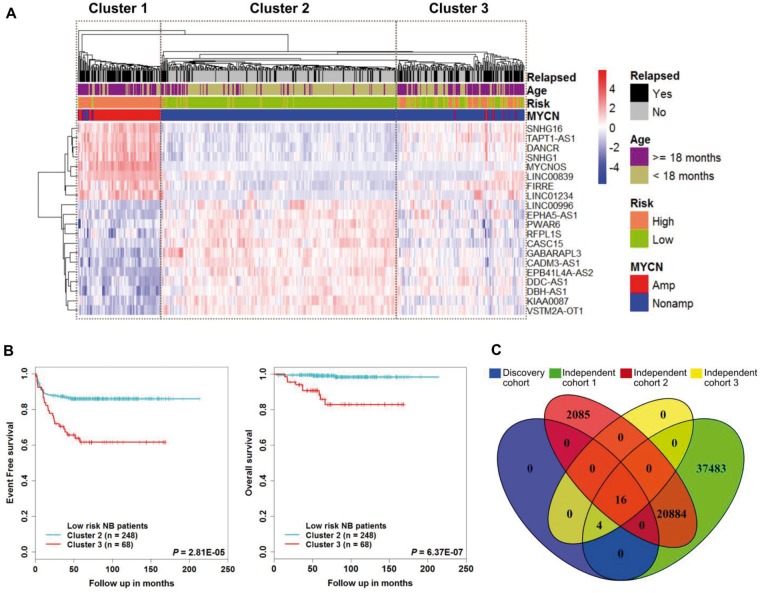
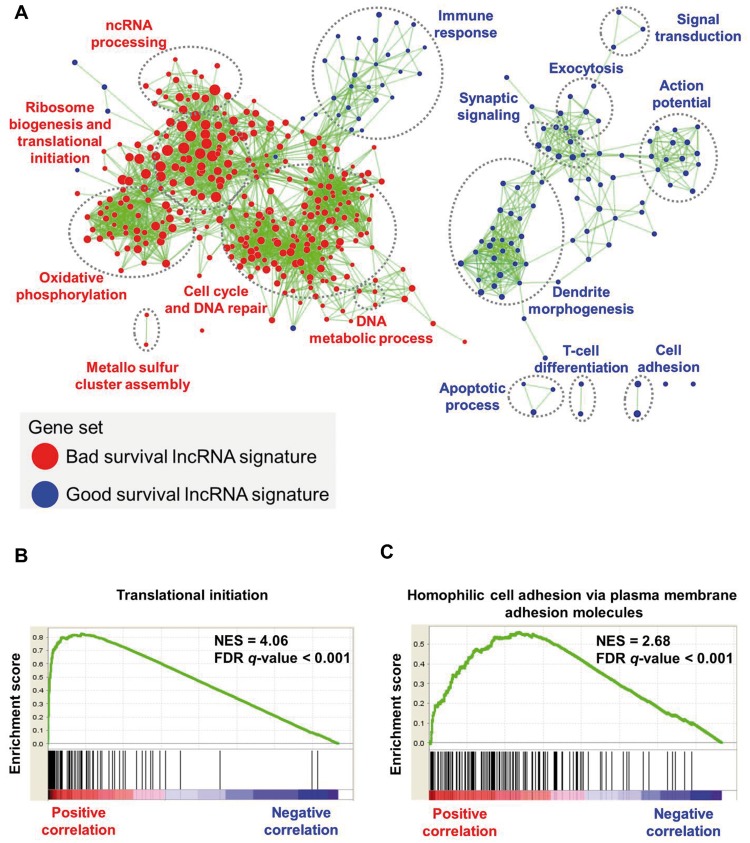
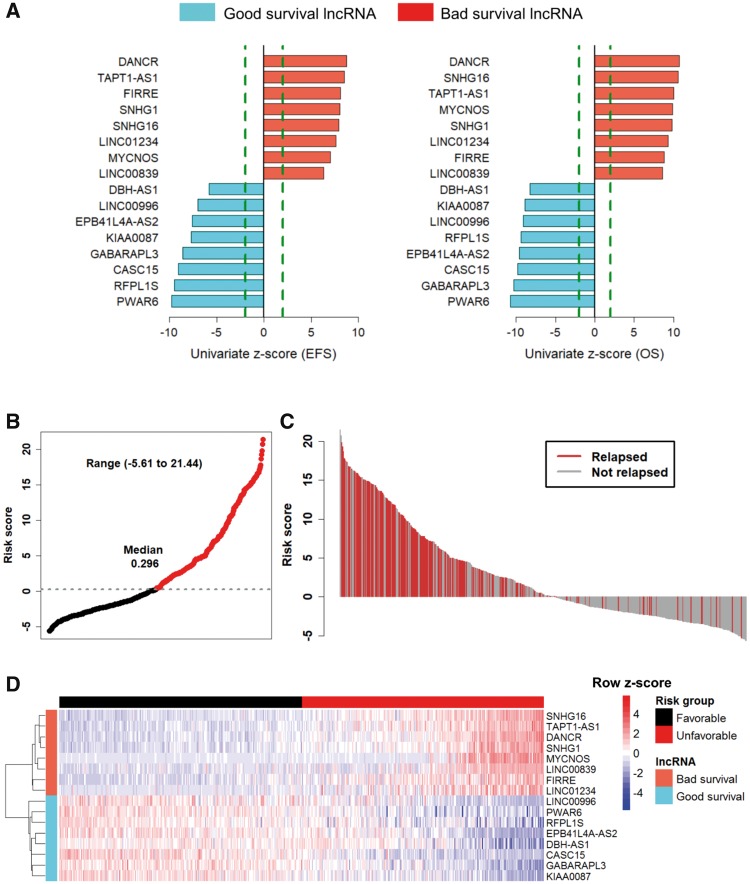
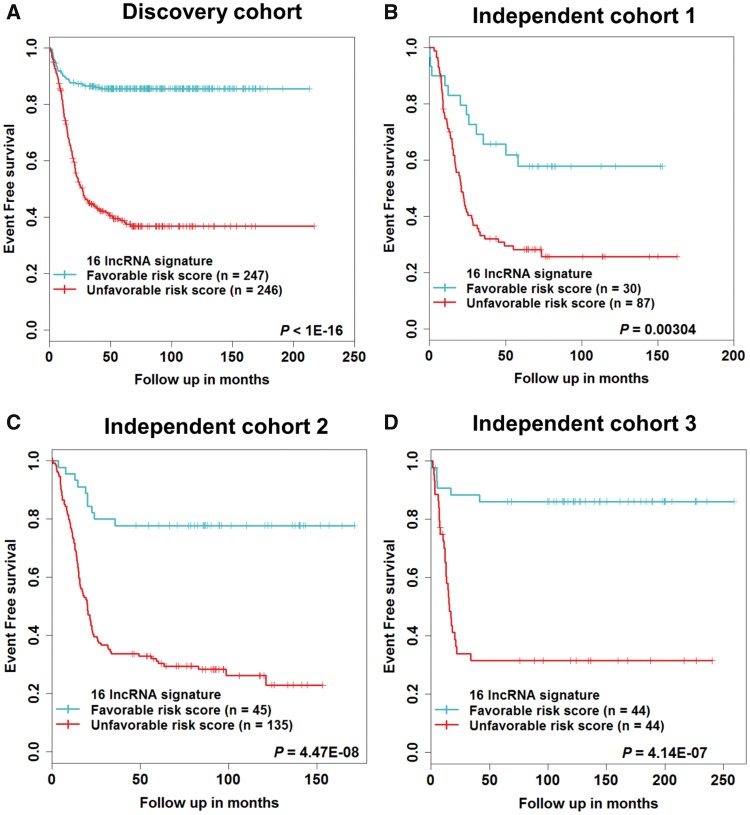
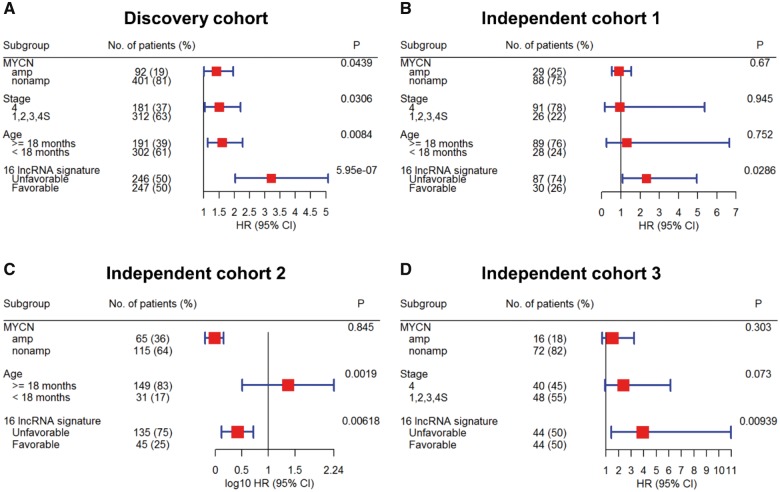
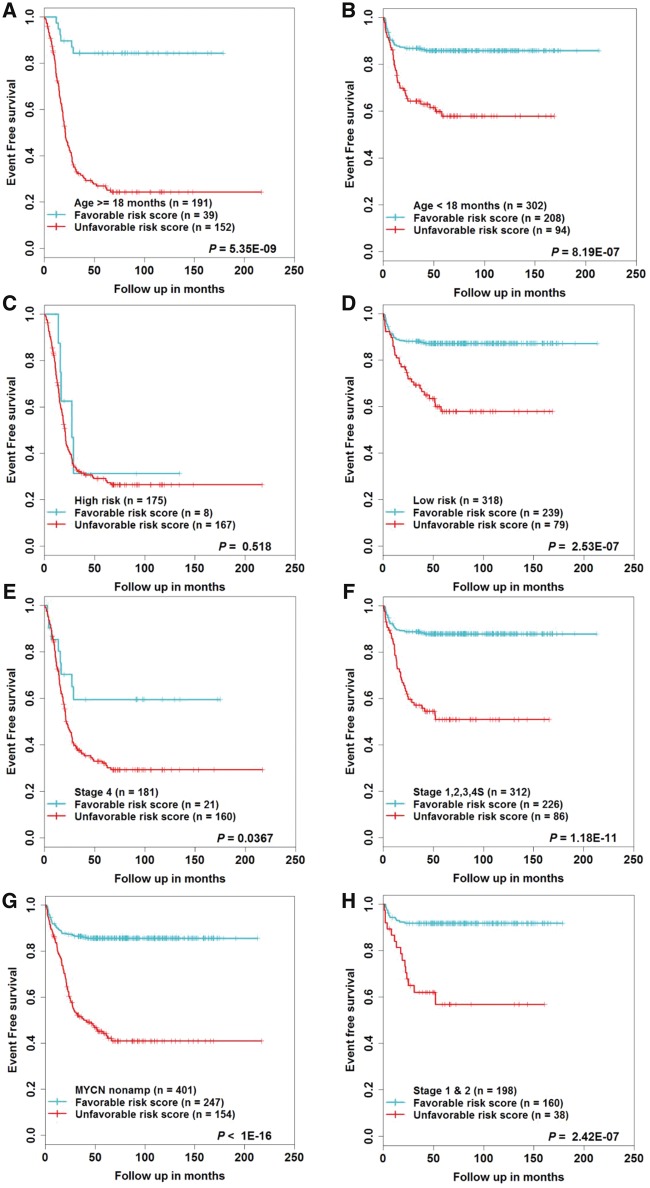
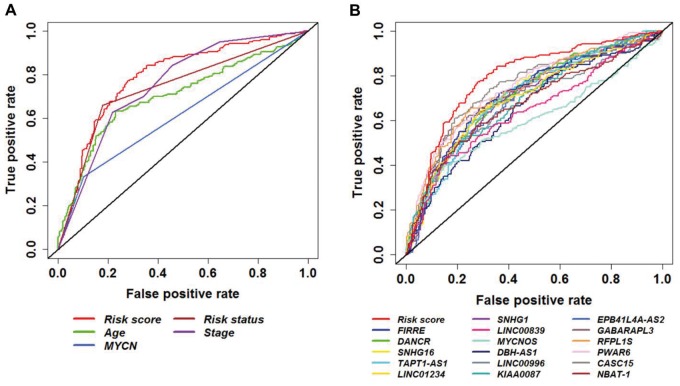
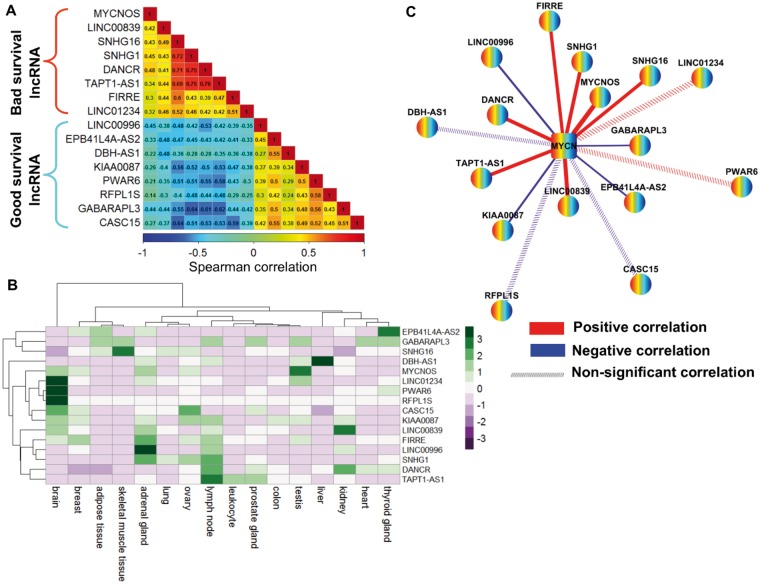
In the discovery cohort, 16 lncRNAs that were differentially expressed (fold change ≥ 2 and adjusted ≤ 0.01) integrated into a prognostic signature. A high risk score group of lncRNA signature had poor event-free survival (EFS; < 1E-16). Notably, lncRNA signature was independent of other clinical risk factors when predicting EFS (hazard ratio = 3.21, = 5.95E-07). The findings were confirmed in independent cohorts ( = 2.86E-02, = 6.18E-03, = 9.39E-03, respectively). Finally, the lncRNA signature had higher accuracy for EFS prediction (area under the curve = 0.788, 95% confidence interval = 0.746 to 0.831).
Here, we report the first (to our knowledge) RNA-seq 16-lncRNA prognostic signature for NB that may contribute to precise clinical stratification and EFS prediction.
目前的临床风险因素可对神经母细胞瘤(NB)患者进行分层以便采取适当治疗,但具有相似临床行为的患者会产生不同的反应。MYCN基因扩增是NB已确定的驱动因素之一,当与高危表现相结合时,会使预后恶化。越来越多的高通量转录组学研究表明,包括NB在内的癌症中存在长链非编码RNA(lncRNA)失调。然而,基于表达的lncRNA特征会因与高危相关的MYCN基因扩增而改变,且对患者预后的预测仍然有限。
我们在一个发现队列(n = 493)中研究了基于RNA测序的lncRNA在MYCN状态和风险状态下的表达谱,并在三个独立队列中进行了验证。在发现队列中,通过单变量Cox回归确定lncRNA的预后相关性,并使用风险评分方法将其整合到一个特征中。制定了一种新的风险评分阈值选择标准,以将患者分层为风险组。使用Kaplan-Meier生存曲线和多变量Cox回归评估风险组和临床亚组的预后。通过受试者工作特征曲线评估lncRNA特征的性能。所有统计检验均为双侧检验。
在发现队列中,16个差异表达的lncRNA(倍数变化≥2且校正P≤0.01)被整合到一个预后特征中。lncRNA特征的高风险评分组无事件生存期(EFS)较差(P<1E-16)。值得注意的是,在预测EFS时,lncRNA特征独立于其他临床风险因素(风险比 = 3.21,P = 5.95E-07)。这些发现在独立队列中得到了证实(P分别为2.86E-02、6.18E-03、9.39E-03)。最后,lncRNA特征对EFS预测具有更高的准确性(曲线下面积 = 0.788,95%置信区间 = 0.746至0.831)。
在此,我们报告了首个(据我们所知)用于NB的基于RNA测序的16-lncRNA预后特征,这可能有助于精确的临床分层和EFS预测。