European Molecular Biology Laboratory, c/o DESY, Notkestrasse 85, 22607 Hamburg, Germany.
Acta Crystallogr D Struct Biol. 2019 Aug 1;75(Pt 8):753-763. doi: 10.1107/S2059798319009392. Epub 2019 Jul 31.
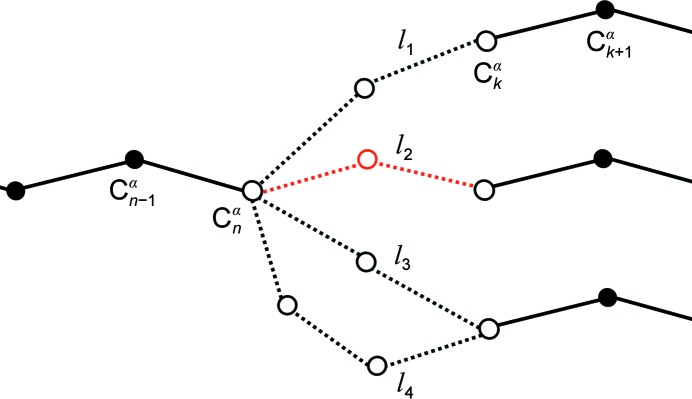
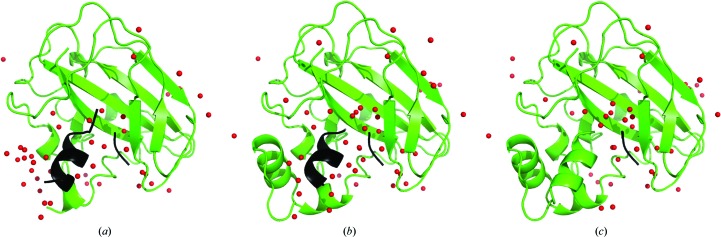
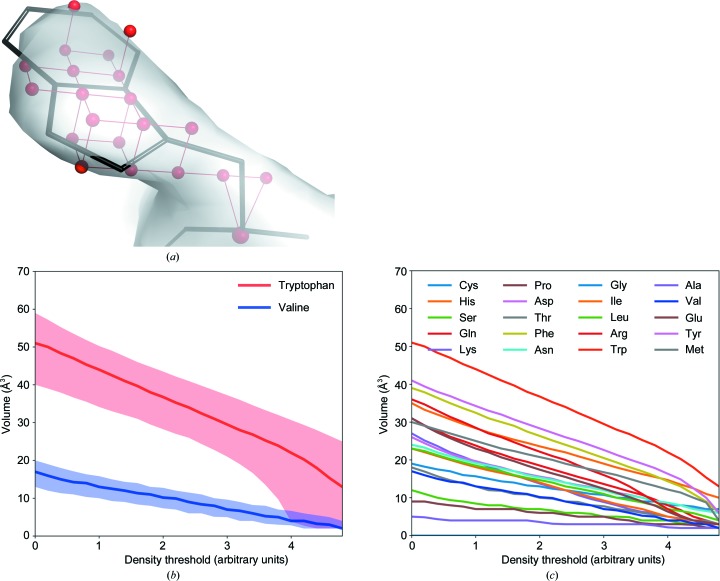
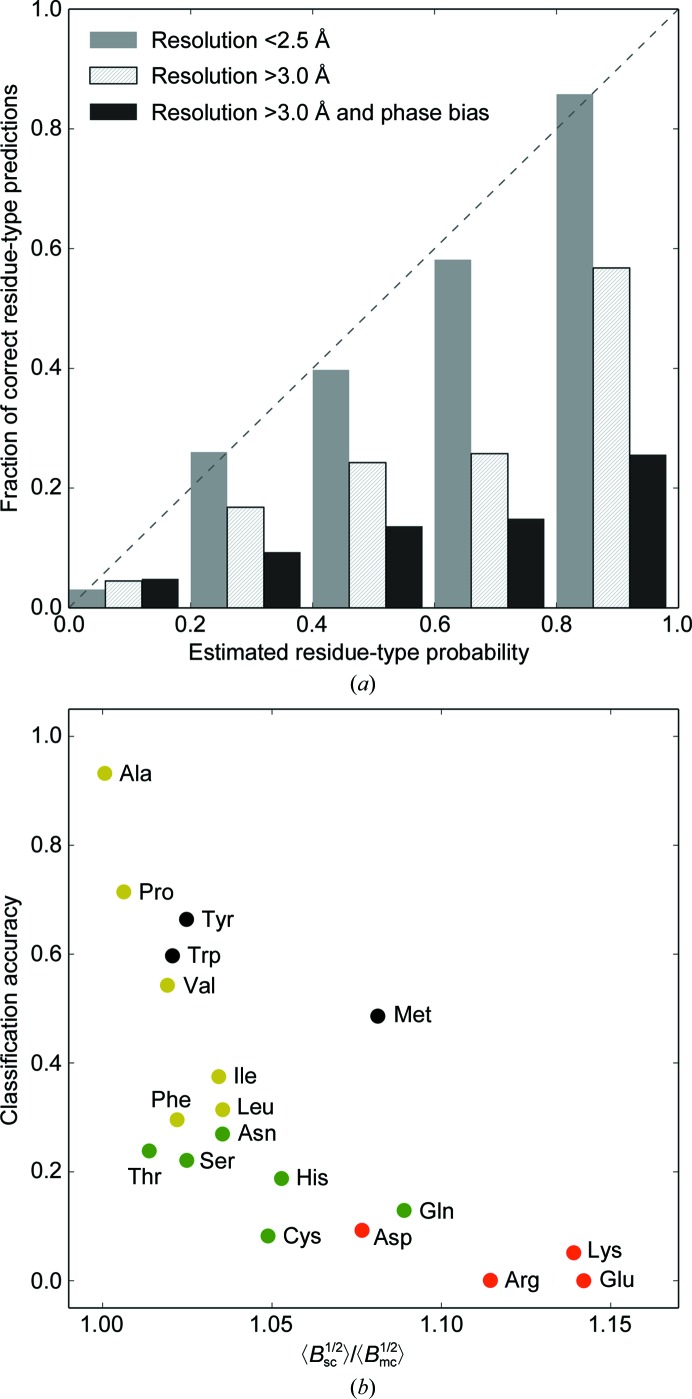
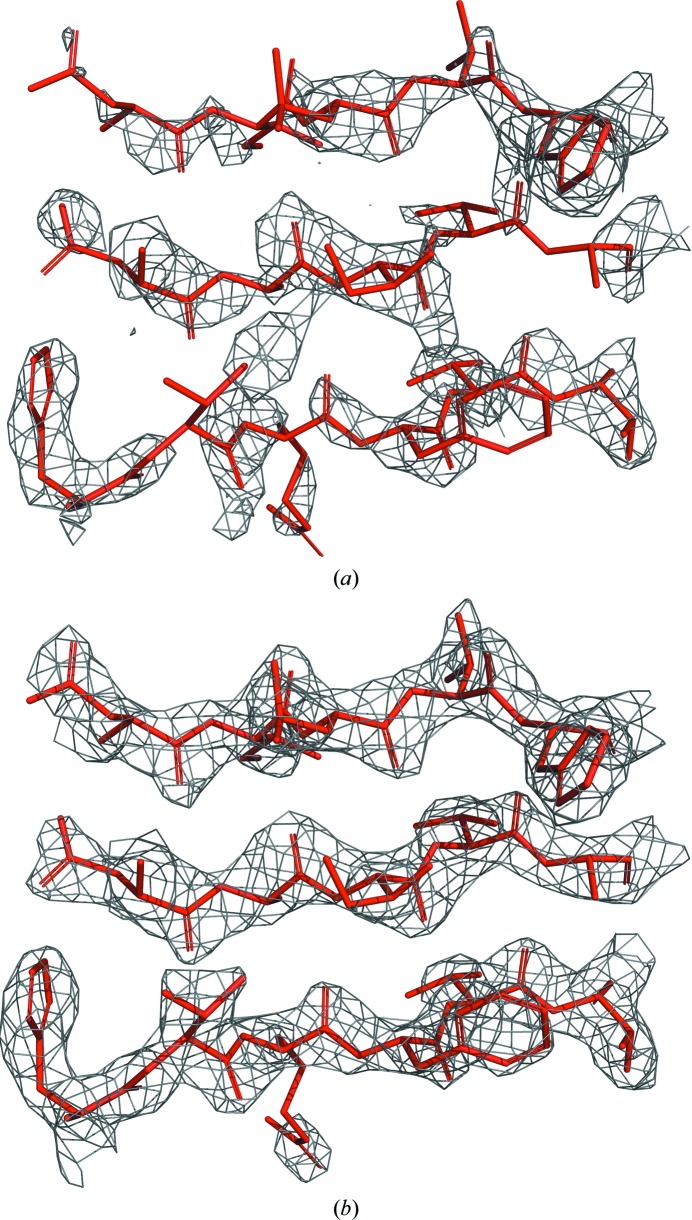
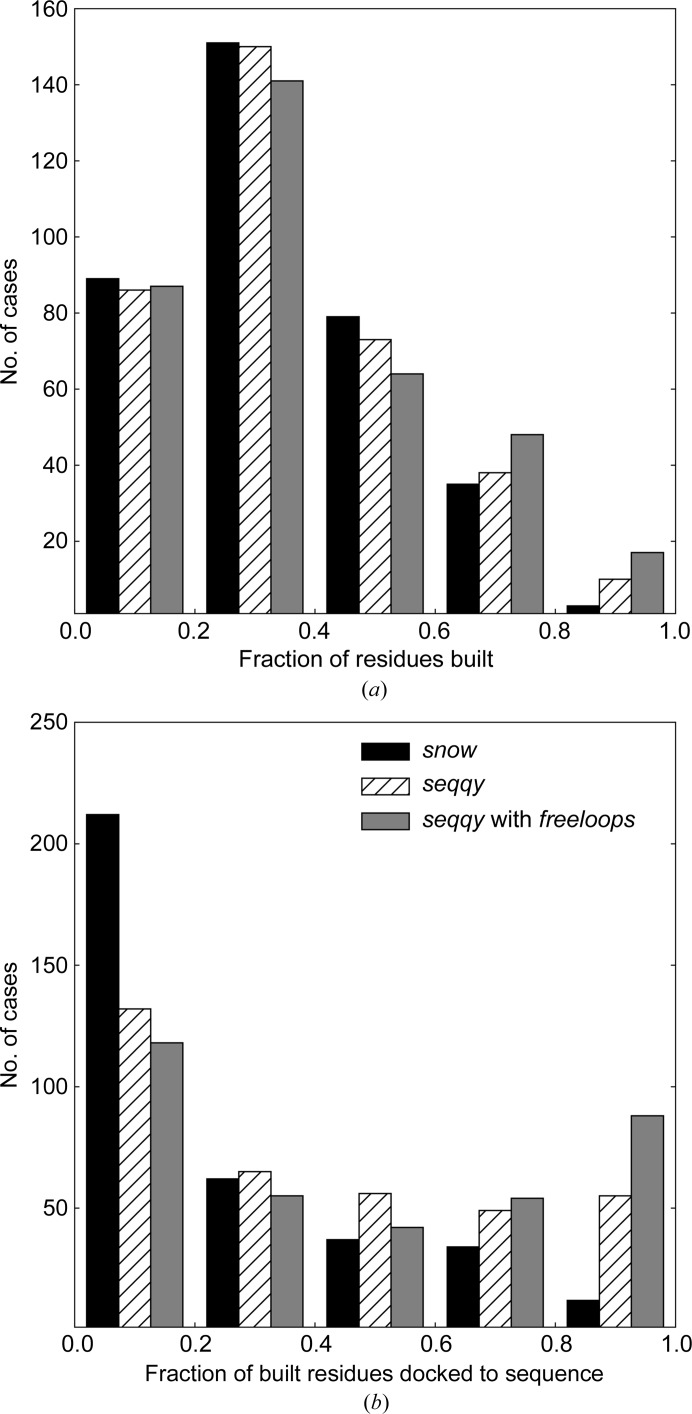
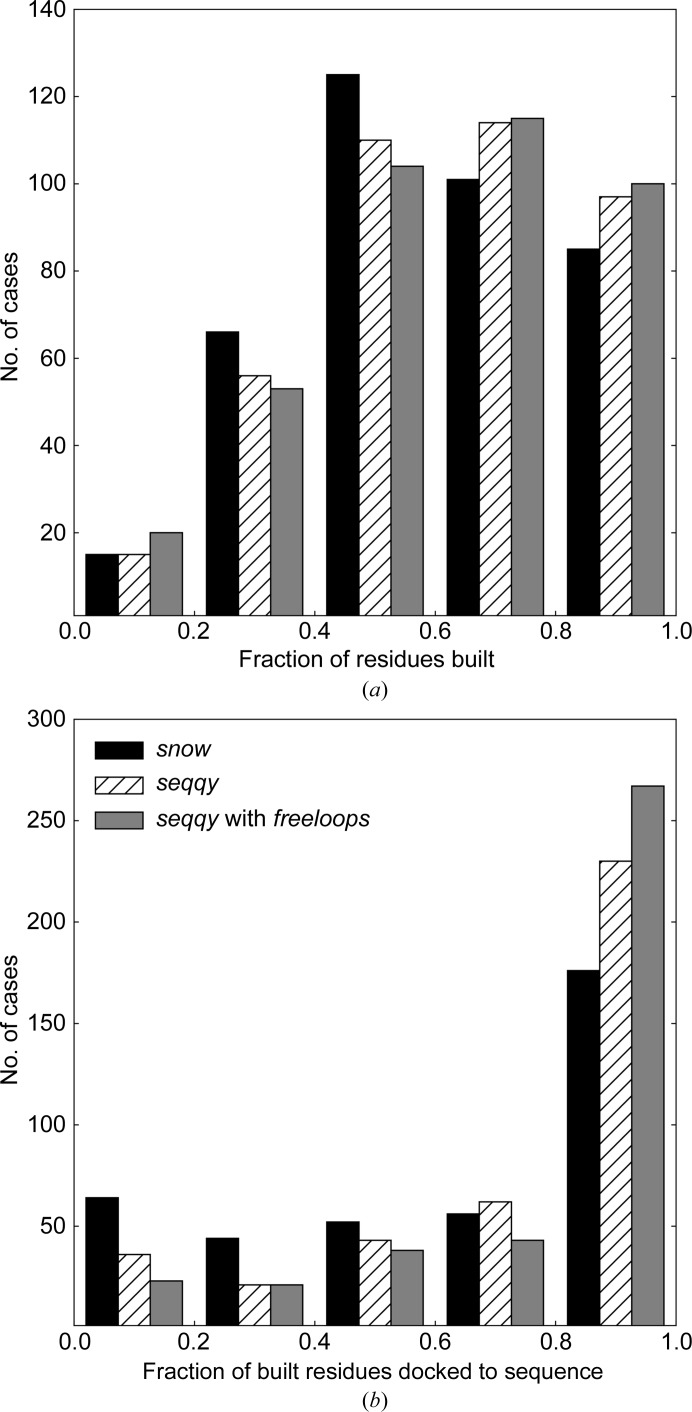
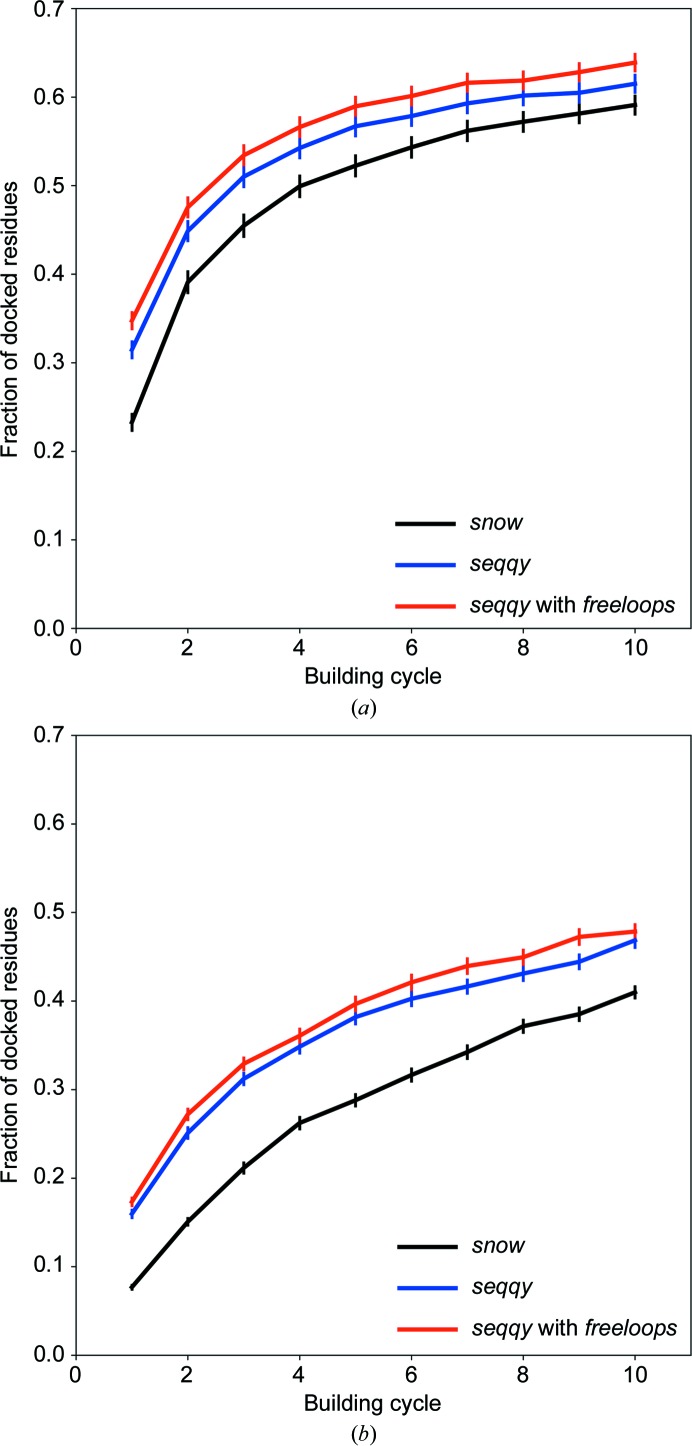
The performance of automated model building in crystal structure determination usually decreases with the resolution of the experimental data, and may result in fragmented models and incorrect side-chain assignment. Presented here are new methods for machine-learning-based docking of main-chain fragments to the sequence and for their sequence-independent connection using a dedicated library of protein fragments. The combined use of these new methods noticeably increases sequence coverage and reduces fragmentation of the protein models automatically built with ARP/wARP.
自动化模型构建在晶体结构测定中的性能通常随实验数据的分辨率降低而下降,并且可能导致模型碎片化和侧链分配不正确。本文介绍了基于机器学习的主链片段对接序列和使用专用蛋白质片段库进行其序列独立连接的新方法。这些新方法的联合使用显著提高了序列覆盖率,并减少了使用 ARP/wARP 自动构建的蛋白质模型的碎片化。