European Molecular Biology Laboratory, c/o DESY, Notkestrasse 85, 22607 Hamburg, Germany.
Acta Crystallogr D Struct Biol. 2020 Mar 1;76(Pt 3):248-260. doi: 10.1107/S2059798320000455. Epub 2020 Feb 28.
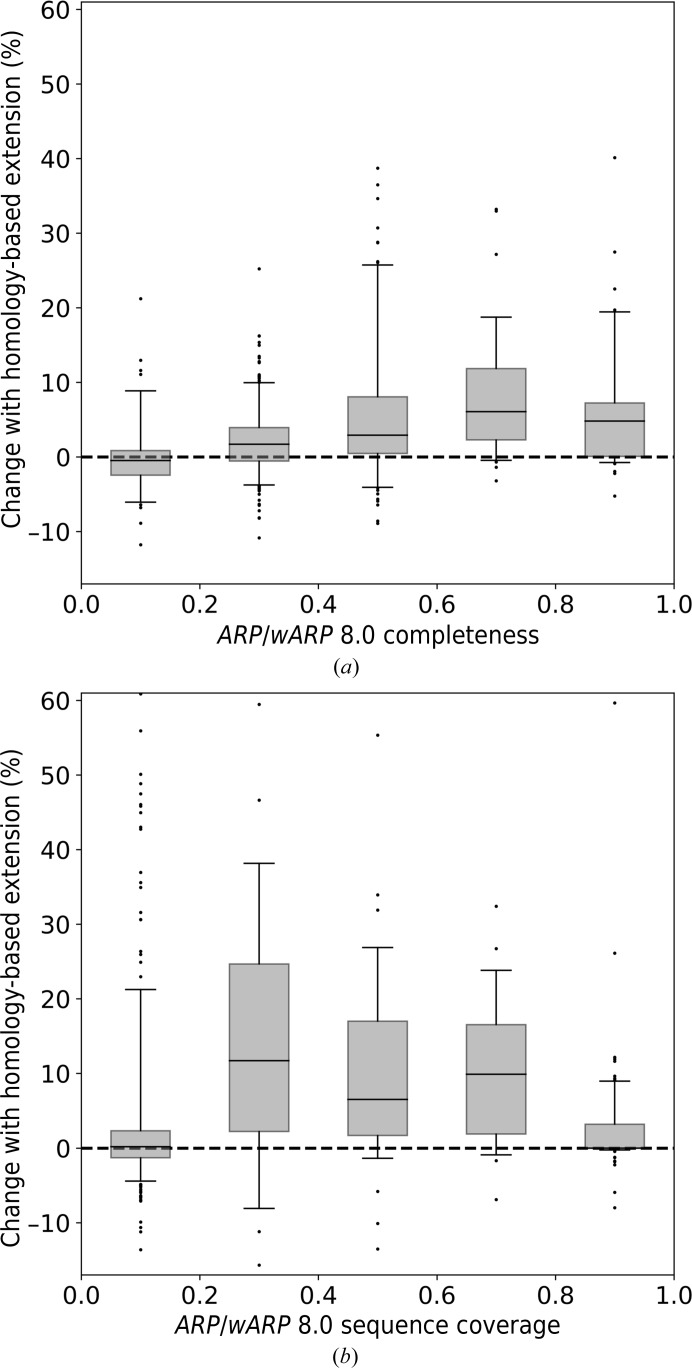
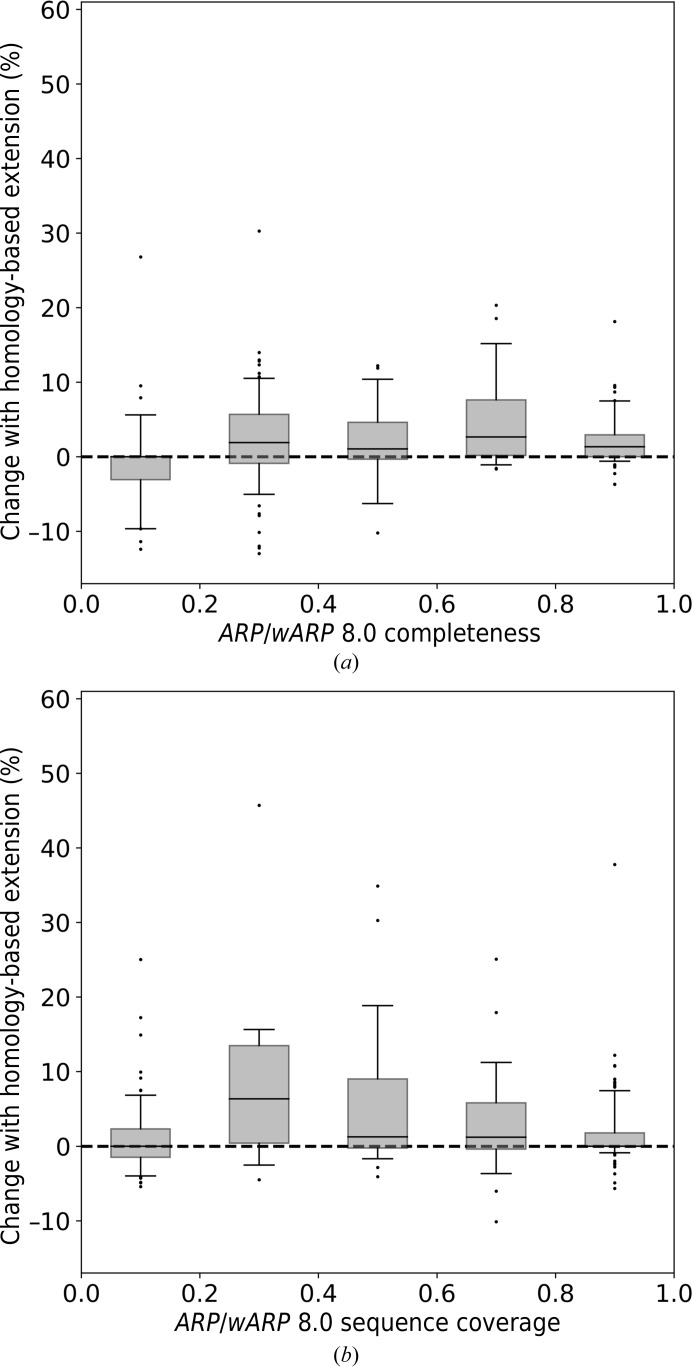
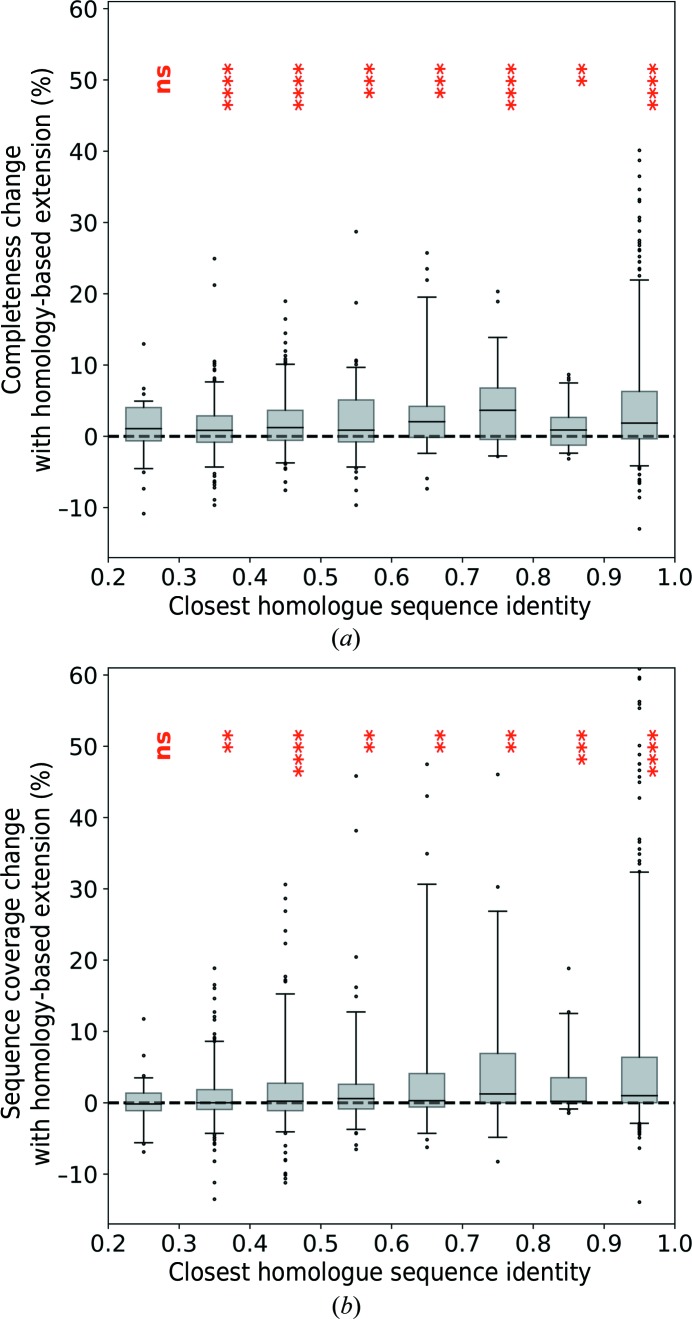
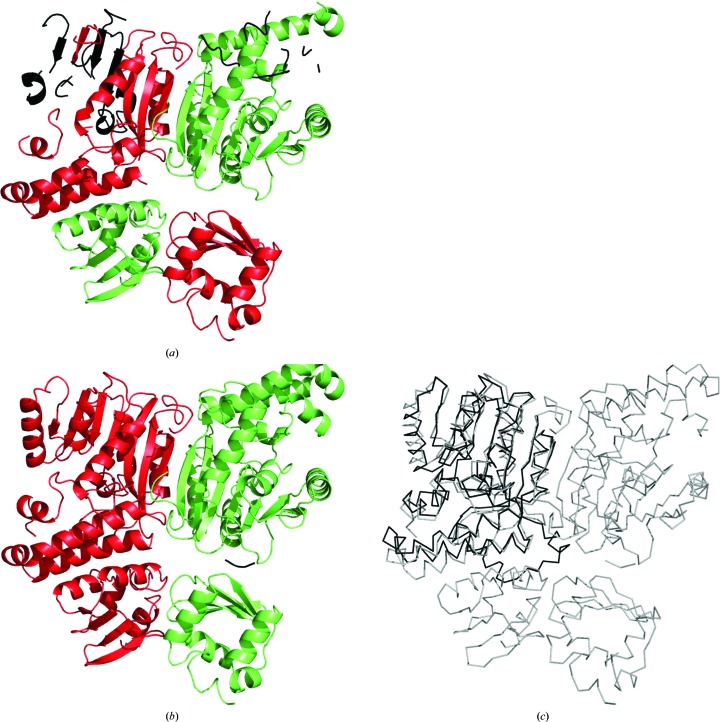
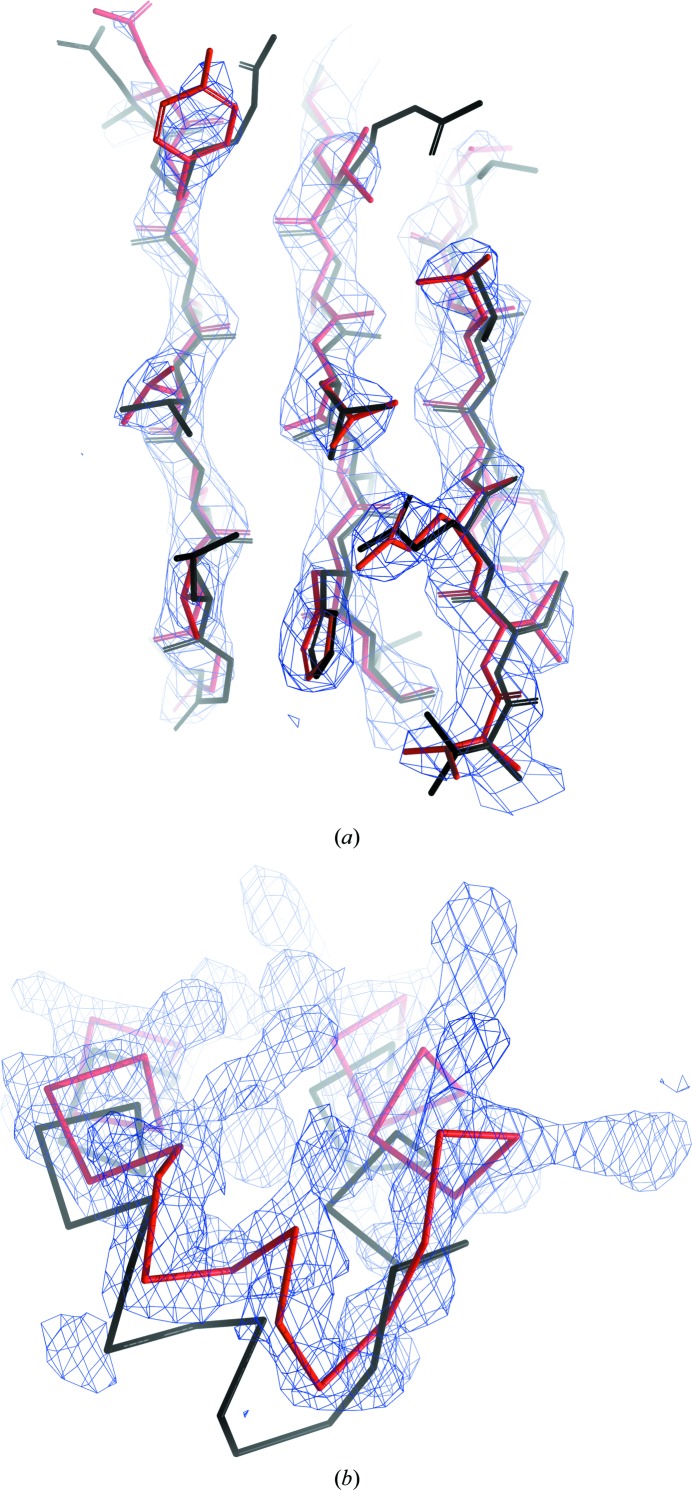
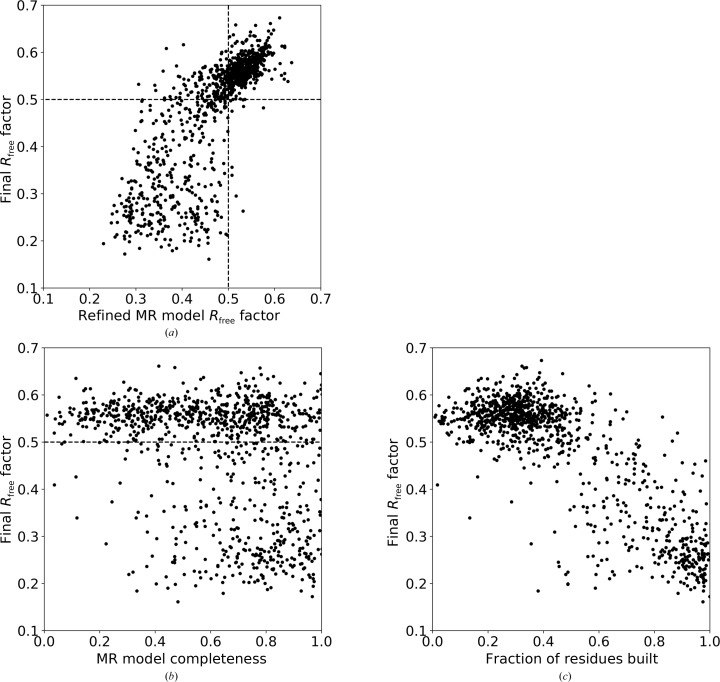
The performance of automated protein model building usually decreases with resolution, mainly owing to the lower information content of the experimental data. This calls for a more elaborate use of the available structural information about macromolecules. Here, a new method is presented that uses structural homologues to improve the quality of protein models automatically constructed using ARP/wARP. The method uses local structural similarity between deposited models and the model being built, and results in longer main-chain fragments that in turn can be more reliably docked to the protein sequence. The application of the homology-based model extension method to the example of a CFA synthase at 2.7 Å resolution resulted in a more complete model with almost all of the residues correctly built and docked to the sequence. The method was also evaluated on 1493 molecular-replacement solutions at a resolution of 4.0 Å and better that were submitted to the ARP/wARP web service for model building. A significant improvement in the completeness and sequence coverage of the built models has been observed.
自动化蛋白质模型构建的性能通常随分辨率降低而下降,主要是由于实验数据的信息量较低。这需要更精细地利用关于大分子的可用结构信息。在这里,提出了一种新的方法,该方法使用结构同源物来改进使用 ARP/wARP 自动构建的蛋白质模型的质量。该方法使用已存储模型和正在构建的模型之间的局部结构相似性,从而产生更长的主链片段,这些片段反过来可以更可靠地对接至蛋白质序列。将基于同源性的模型扩展方法应用于分辨率为 2.7 Å 的 CFA 合酶的示例中,得到了一个更完整的模型,几乎所有的残基都正确构建并对接至序列。该方法还在分辨率为 4.0 Å 及更好的 1493 个分子置换解决方案上进行了评估,这些解决方案已提交给 ARP/wARP 网络服务进行模型构建。已观察到构建模型的完整性和序列覆盖率有显著提高。