Department of Emergency and Chest Pain Center, Qilu Hospital of Shandong University, Jinan, China.
Clinical Research Center for Emergency and Critical Care Medicine of Shandong Province, Institute of Emergency and Critical Care Medicine of Shandong University, Qilu Hospital of Shandong University, Jinan, China.
J Cell Mol Med. 2019 Oct;23(10):6897-6906. doi: 10.1111/jcmm.14573. Epub 2019 Aug 5.
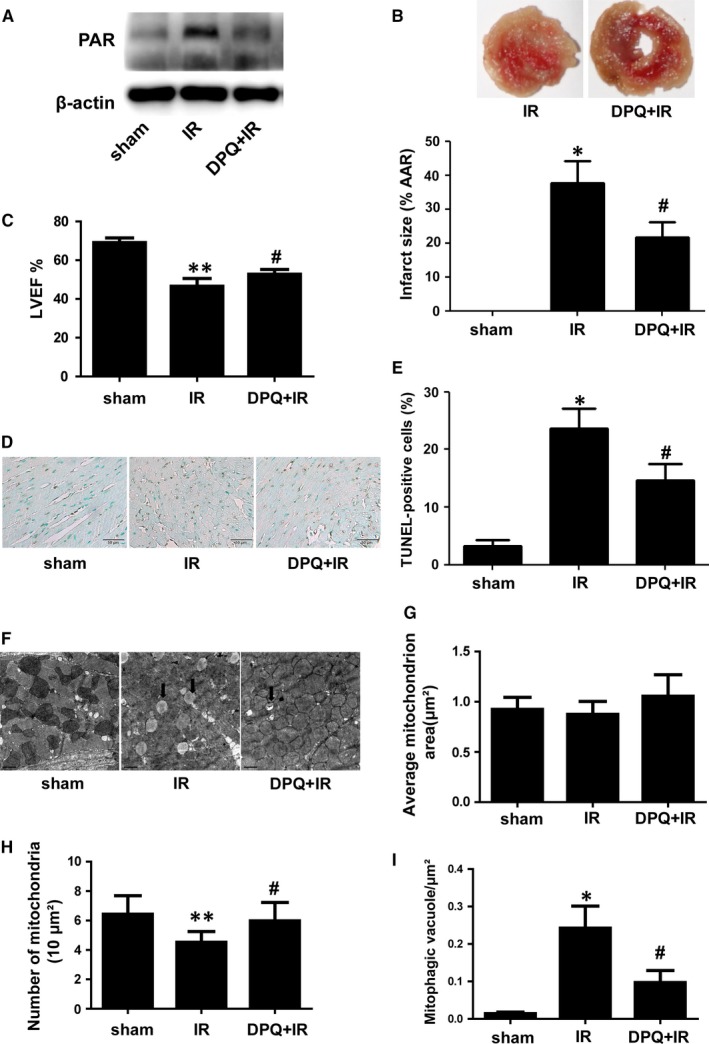
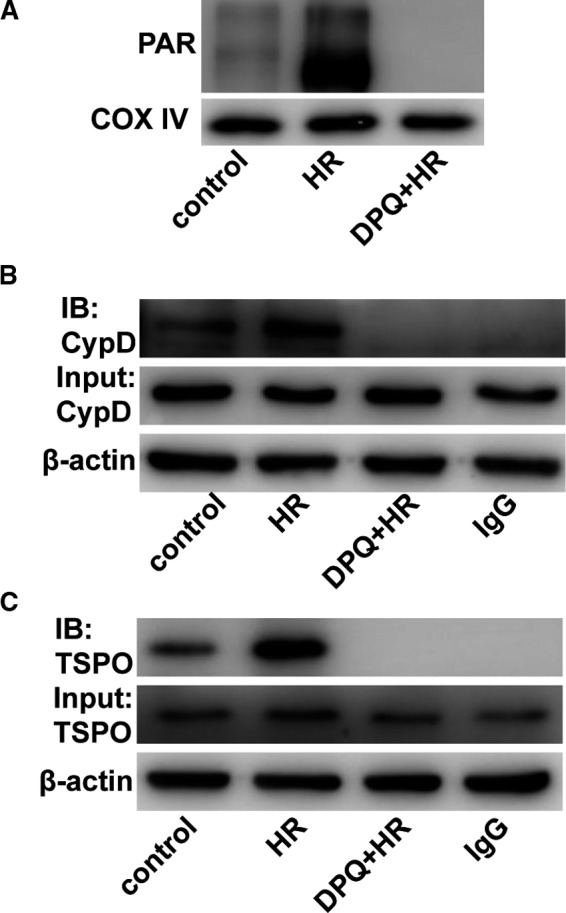
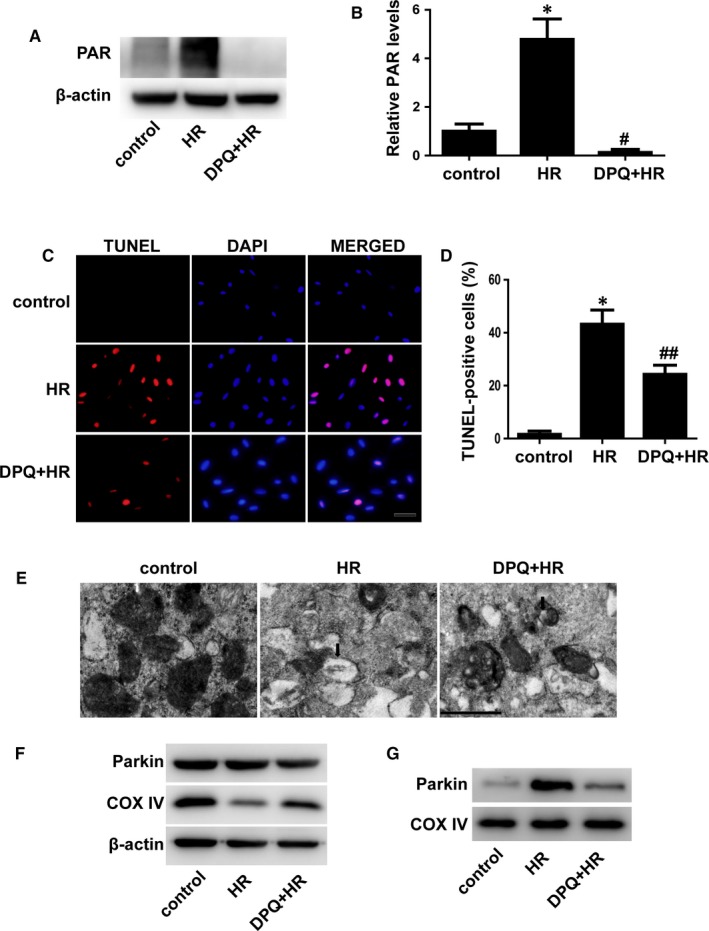
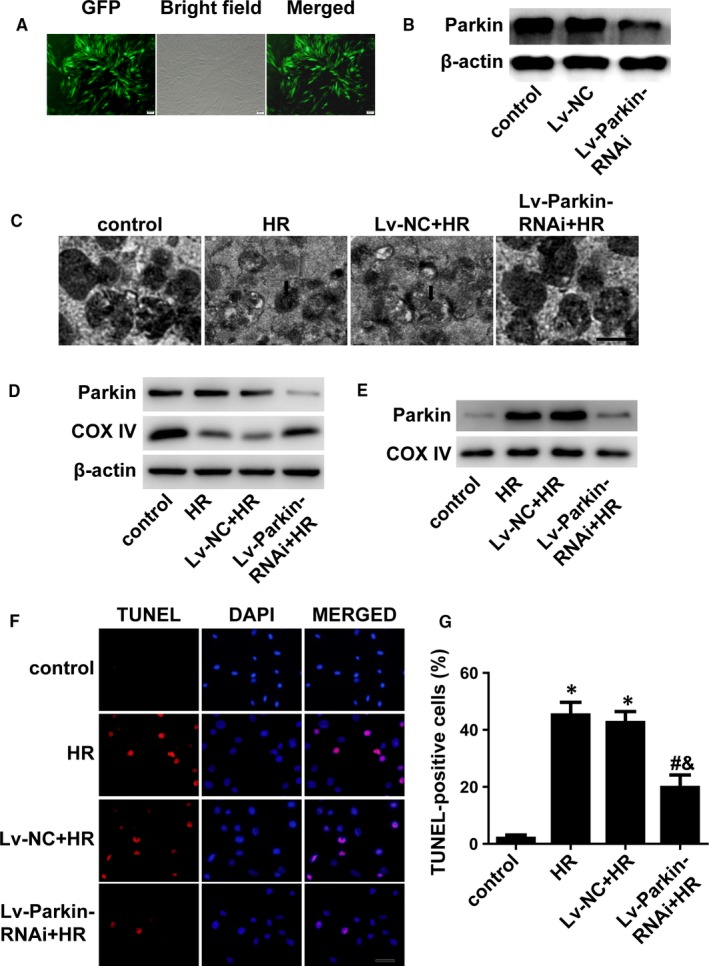
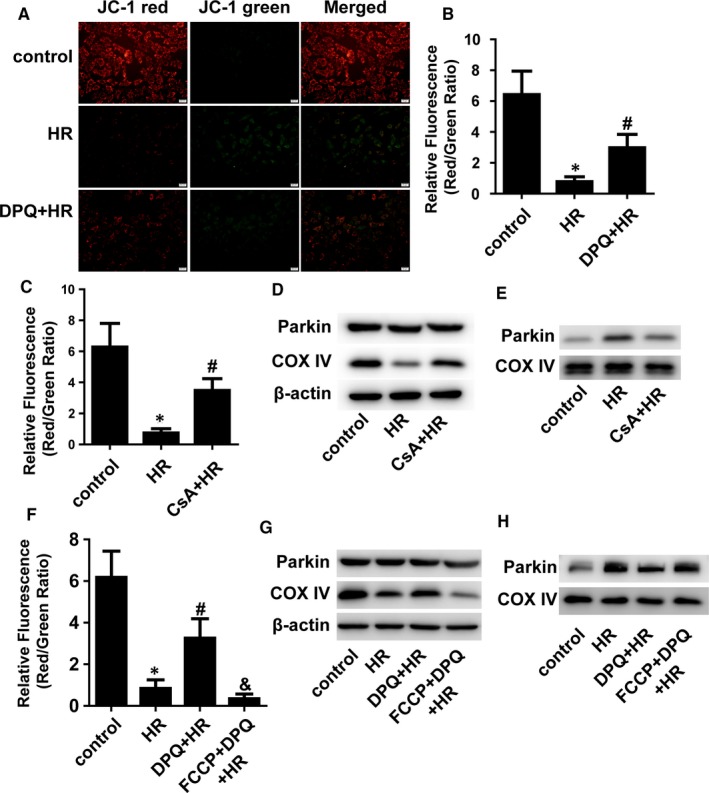
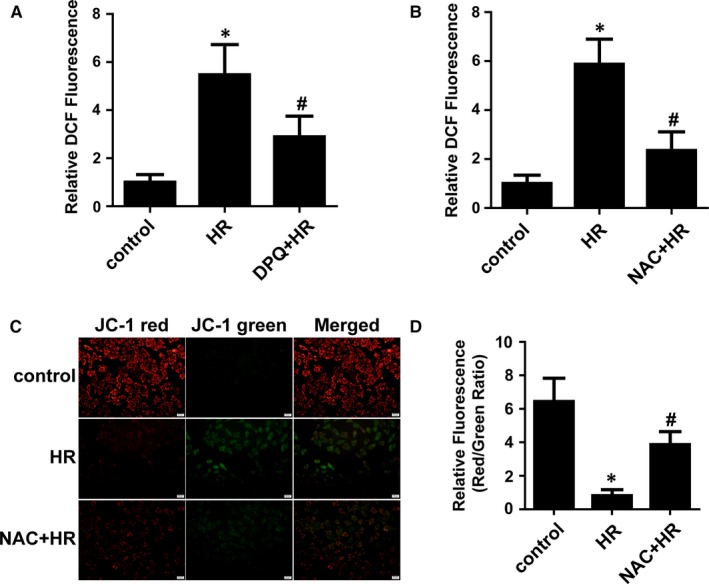
Myocardial ischaemia/reperfusion (I/R) injury attenuates the beneficial effects of reperfusion therapy. Poly(ADP-ribose) polymerase (PARP) is overactivated during myocardial I/R injury. Mitophagy plays a critical role in the development of myocardial I/R injury. However, the effect of PARP activation on mitophagy in cardiomyocytes is unknown. In this study, we found that I/R induced PARP activation and mitophagy in mouse hearts. Poly(ADP-ribose) polymerase inhibition reduced the infarct size and suppressed mitophagy after myocardial I/R injury. In vitro, hypoxia/reoxygenation (H/R) activated PARP, promoted mitophagy and induced cell apoptosis in cardiomyocytes. Poly(ADP-ribose) polymerase inhibition suppressed H/R-induced mitophagy and cell apoptosis. Parkin knockdown with lentivirus vectors inhibited mitophagy and prevented cell apoptosis in H/R-treated cells. Poly(ADP-ribose) polymerase inhibition prevented the loss of the mitochondrial membrane potential (ΔΨm). Cyclosporin A maintained ΔΨm and suppressed mitophagy but FCCP reduced the effect of PARP inhibition on ΔΨm and promoted mitophagy, indicating the critical role of ΔΨm in H/R-induced mitophagy. Furthermore, reactive oxygen species (ROS) and poly(ADP-ribosylation) of CypD and TSPO might contribute to the regulation of ΔΨm by PARP. Our findings thus suggest that PARP inhibition protects against I/R-induced cell apoptosis by suppressing excessive mitophagy via the ΔΨm/Parkin pathway.
心肌缺血/再灌注(I/R)损伤会减弱再灌注治疗的有益作用。多聚(ADP-核糖)聚合酶(PARP)在心肌 I/R 损伤过程中过度激活。自噬在心肌 I/R 损伤的发生发展中起着关键作用。然而,PARP 激活对心肌细胞自噬的影响尚不清楚。在本研究中,我们发现 I/R 诱导了小鼠心脏中的 PARP 激活和自噬。PARP 抑制减少了心肌 I/R 损伤后的梗死面积并抑制了自噬。在体外,缺氧/复氧(H/R)激活了 PARP,促进了心肌细胞的自噬并诱导了细胞凋亡。PARP 抑制抑制了 H/R 诱导的自噬和细胞凋亡。用慢病毒载体敲低 Parkin 抑制了 H/R 处理细胞中的自噬并阻止了细胞凋亡。PARP 抑制防止了线粒体膜电位(ΔΨm)的丧失。环孢素 A 维持了 ΔΨm 并抑制了自噬,但 FCCP 降低了 PARP 抑制对 ΔΨm 的作用并促进了自噬,表明 ΔΨm 在 H/R 诱导的自噬中起关键作用。此外,CypD 和 TSPO 的活性氧(ROS)和多聚(ADP-核糖基化)可能有助于 PARP 调节 ΔΨm。因此,我们的研究结果表明,PARP 抑制通过抑制过度的自噬来保护心脏免受 I/R 诱导的细胞凋亡,这种作用是通过 ΔΨm/Parkin 通路实现的。