Jonas Eric, Kuhn Stefan
Department of Computer Science, University of Chicago, Chicago, USA.
School of Computer Science and Informatics, Leicester, UK.
J Cheminform. 2019 Aug 6;11(1):50. doi: 10.1186/s13321-019-0374-3.
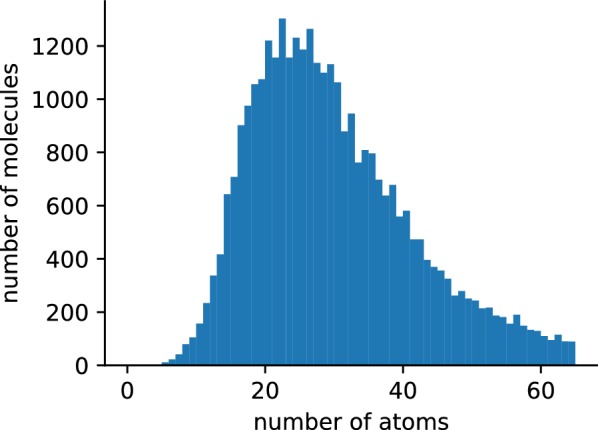
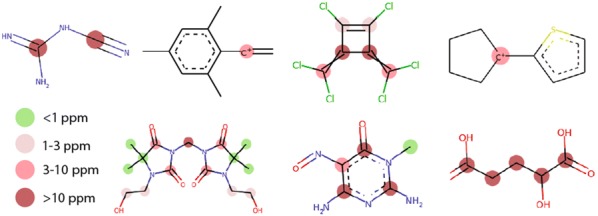
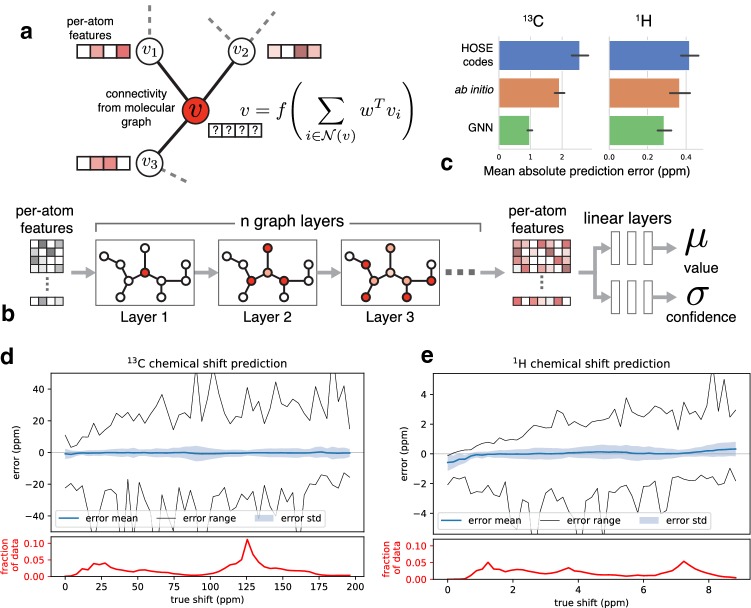
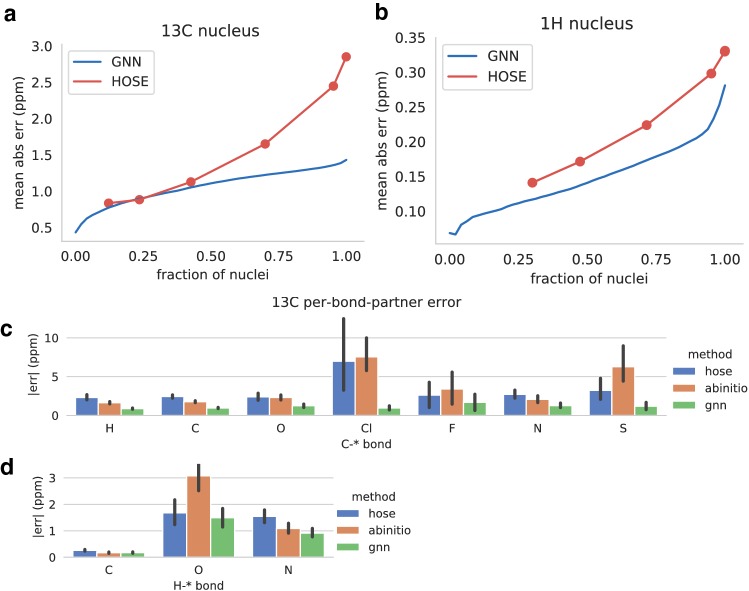
Accurate calculation of specific spectral properties for NMR is an important step for molecular structure elucidation. Here we report the development of a novel machine learning technique for accurately predicting chemical shifts of both [Formula: see text] and [Formula: see text] nuclei which exceeds DFT-accessible accuracy for [Formula: see text] and [Formula: see text] for a subset of nuclei, while being orders of magnitude more performant. Our method produces estimates of uncertainty, allowing for robust and confident predictions, and suggests future avenues for improved performance.
准确计算核磁共振(NMR)的特定光谱特性是阐明分子结构的重要一步。在此,我们报告了一种新型机器学习技术的开发,该技术可精确预测[化学式:见原文]和[化学式:见原文]原子核的化学位移,对于一部分原子核,其预测精度超过了密度泛函理论(DFT)可达的精度,且在[化学式:见原文]和[化学式:见原文]方面比DFT高出几个数量级。我们的方法能够生成不确定性估计值,从而实现稳健且可靠的预测,并为性能改进指明了未来的方向。