Eck Institute for Global Health & Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556.
Infectious Disease and Microbiome Program, Broad Institute, Cambridge, MA 02142.
G3 (Bethesda). 2019 Oct 7;9(10):3249-3262. doi: 10.1534/g3.119.400445.
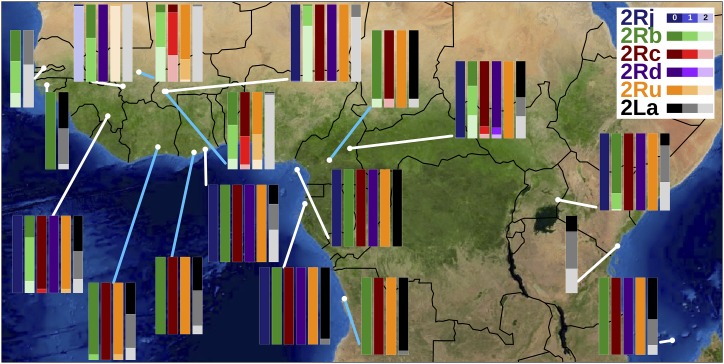
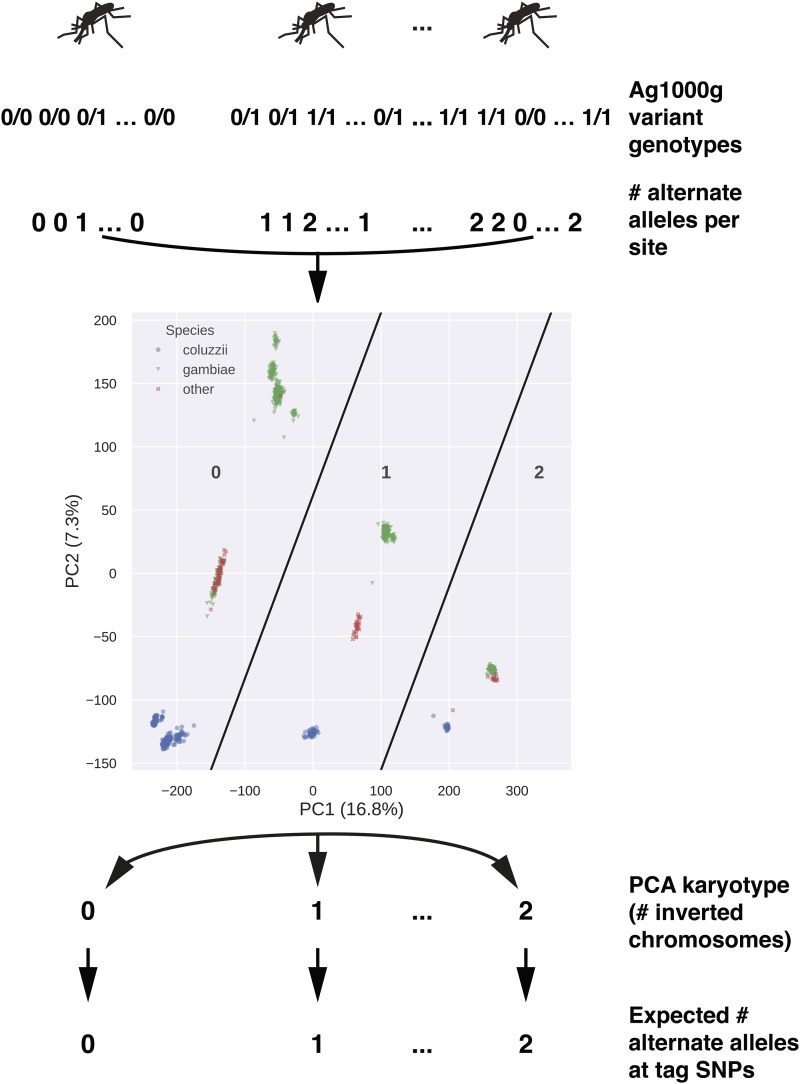
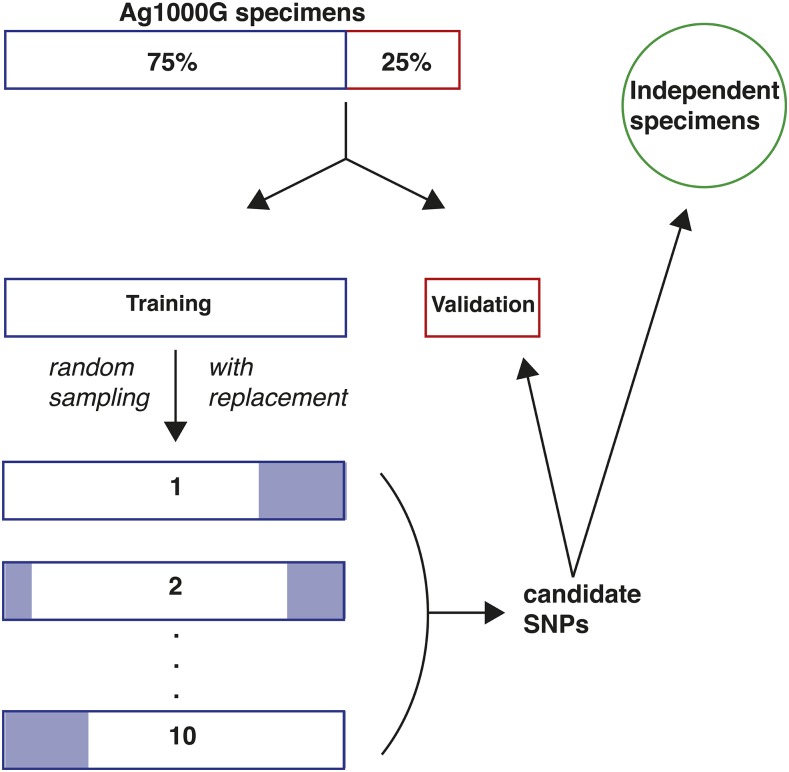
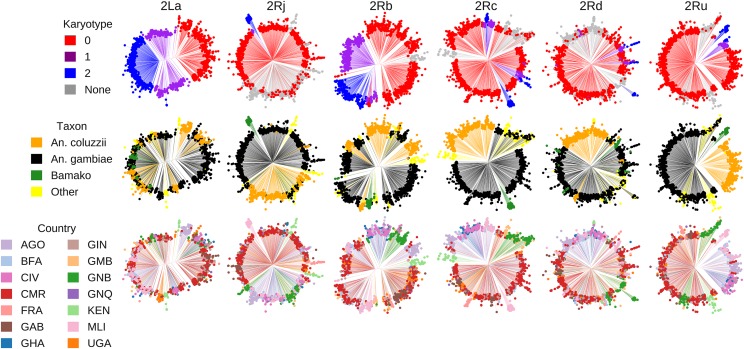
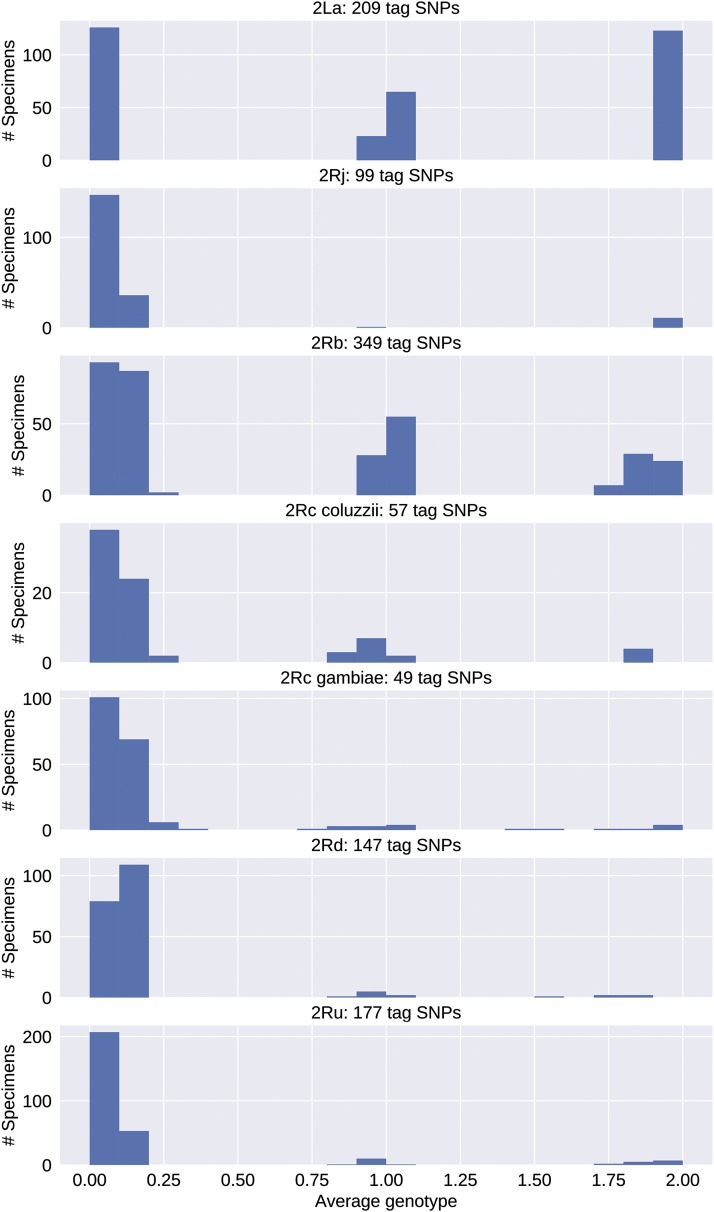
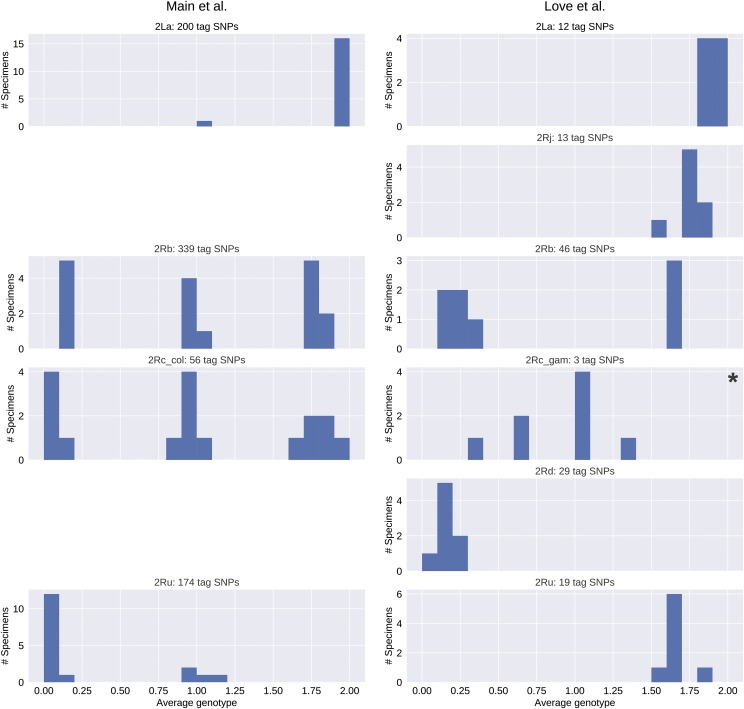
Chromosomal inversion polymorphisms play an important role in adaptation to environmental heterogeneities. For mosquito species in the complex that are significant vectors of human malaria, paracentric inversion polymorphisms are abundant and are associated with ecologically and epidemiologically important phenotypes. Improved understanding of these traits relies on determining mosquito karyotype, which currently depends upon laborious cytogenetic methods whose application is limited both by the requirement for specialized expertise and for properly preserved adult females at specific gonotrophic stages. To overcome this limitation, we developed sets of tag single nucleotide polymorphisms (SNPs) inside inversions whose biallelic genotype is strongly correlated with inversion genotype. We leveraged 1,347 fully sequenced and genomes in the Ag1000G database of natural variation. Beginning with principal components analysis (PCA) of population samples, applied to windows of the genome containing individual chromosomal rearrangements, we classified samples into three inversion genotypes, distinguishing homozygous inverted and homozygous uninverted groups by inclusion of the small subset of specimens in Ag1000G that are associated with cytogenetic metadata. We then assessed the correlation between candidate tag SNP genotypes and PCA-based inversion genotypes in our training sets, selecting those candidates with >80% agreement. Our initial tests both in held-back validation samples from Ag1000G and in data independent of Ag1000G suggest that when used for inversion genotyping of sequenced mosquitoes, these tags perform better than traditional cytogenetics, even for specimens where only a small subset of the tag SNPs can be successfully ascertained.
染色体倒位多态性在适应环境异质性方面起着重要作用。对于作为人类疟疾重要传播媒介的复杂蚊种,旁侧倒位多态性非常丰富,与具有生态和流行病学重要意义的表型相关。对这些特征的深入了解依赖于确定蚊子的核型,而这目前依赖于繁琐的细胞遗传学方法,其应用受到专门知识的要求以及特定生殖阶段的适当保存的成年雌性的限制。为了克服这一限制,我们在倒位内部开发了一组标签单核苷酸多态性(SNP),其双等位基因基因型与倒位基因型强烈相关。我们利用了 1347 个在自然变异的 Ag1000G 数据库中完全测序的基因组。从包含个体染色体重排的基因组窗口的群体样本主成分分析(PCA)开始,我们将样本分为三种倒位基因型,通过包括 Ag1000G 中与细胞遗传学元数据相关的小部分标本,将同型倒位和同型未倒位的群体区分开来。然后,我们评估了候选标记 SNP 基因型与 PCA 基于的我们训练集的倒位基因型之间的相关性,选择那些具有 >80%一致性的候选者。我们在 Ag1000G 的保留验证样本和独立于 Ag1000G 的数据中的初步测试表明,当用于测序蚊子的 倒位基因分型时,这些标记的性能优于传统细胞遗传学,即使对于只有一小部分标记 SNP 可以成功确定的标本也是如此。