Hubei Key Laboratory of Agricultural Bioinformatics, College of Informatics, Huazhong Agricultural University, Wuhan 430070, P.R. China.
National Institute of Genomics and Advanced Biotechnology, National Agriculture Research Center, Islamabad 44051, Pakistan.
Nucleic Acids Res. 2020 Jan 8;48(D1):D956-D963. doi: 10.1093/nar/gkz711.
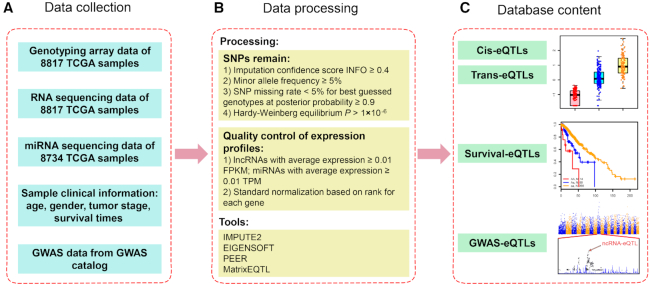
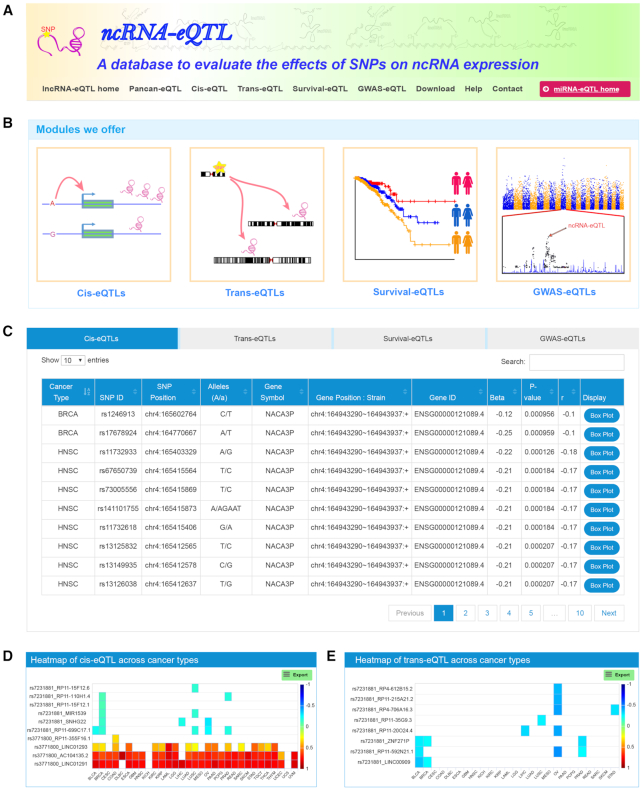
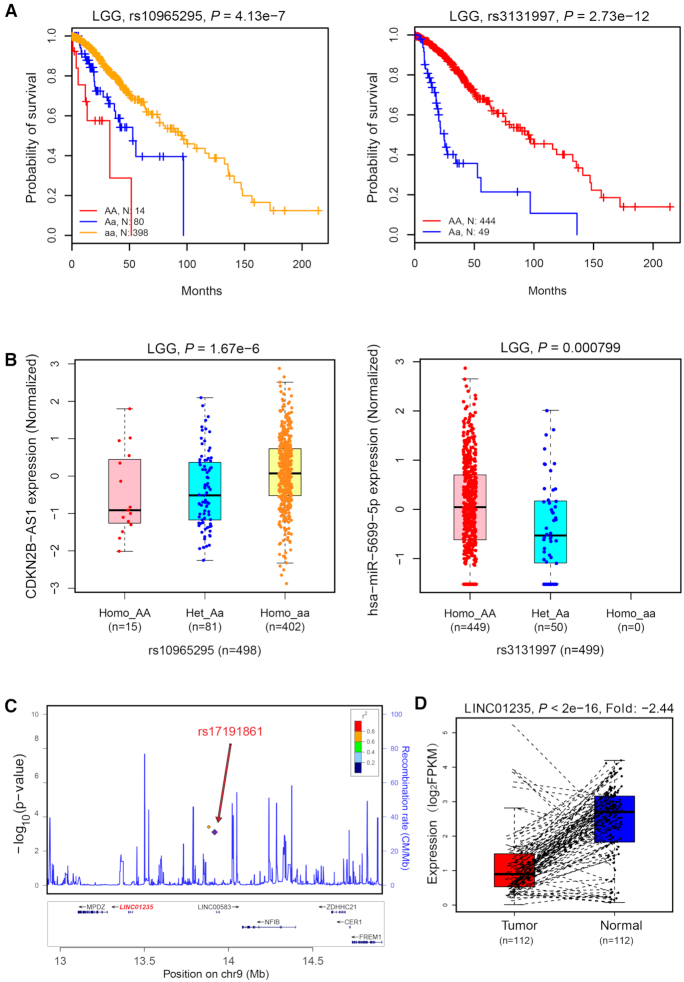
Numerous studies indicate that non-coding RNAs (ncRNAs) have critical functions across biological processes, and single-nucleotide polymorphisms (SNPs) could contribute to diseases or traits through influencing ncRNA expression. However, the associations between SNPs and ncRNA expression are largely unknown. Therefore, genome-wide expression quantitative trait loci (eQTL) analysis to assess the effects of SNPs on ncRNA expression, especially in multiple cancer types, will help to understand how risk alleles contribute toward tumorigenesis and cancer development. Using genotype data and expression profiles of ncRNAs of >8700 samples from The Cancer Genome Atlas (TCGA), we developed a computational pipeline to systematically identify ncRNA-related eQTLs (ncRNA-eQTLs) across 33 cancer types. We identified a total of 6 133 278 and 721 122 eQTL-ncRNA pairs in cis-eQTL and trans-eQTL analyses, respectively. Further survival analyses identified 8312 eQTLs associated with patient survival times. Furthermore, we linked ncRNA-eQTLs to genome-wide association study (GWAS) data and found 262 332 ncRNA-eQTLs overlapping with known disease- and trait-associated loci. Finally, a user-friendly database, ncRNA-eQTL (http://ibi.hzau.edu.cn/ncRNA-eQTL), was developed for free searching, browsing and downloading of all ncRNA-eQTLs. We anticipate that such an integrative and comprehensive resource will improve our understanding of the mechanistic basis of human complex phenotypic variation, especially for ncRNA- and cancer-related studies.
大量研究表明,非编码 RNA(ncRNA)在生物过程中具有关键功能,单核苷酸多态性(SNP)可通过影响 ncRNA 表达而导致疾病或表型。然而,SNP 与 ncRNA 表达之间的关联在很大程度上尚不清楚。因此,进行全基因组表达数量性状基因座(eQTL)分析以评估 SNP 对 ncRNA 表达的影响,特别是在多种癌症类型中,将有助于了解风险等位基因如何导致肿瘤发生和癌症发展。我们使用来自癌症基因组图谱(TCGA)的>8700 个样本的基因型数据和 ncRNA 表达谱,开发了一种计算流程来系统地识别 33 种癌症类型中的 ncRNA 相关 eQTL(ncRNA-eQTL)。在顺式-eQTL 和反式-eQTL 分析中,我们分别鉴定出了总共 6133278 对和 721122 对 eQTL-ncRNA 对。进一步的生存分析确定了与患者生存时间相关的 8312 个 eQTL。此外,我们将 ncRNA-eQTL 与全基因组关联研究(GWAS)数据相关联,并发现了 262332 个与已知疾病和性状相关的位点重叠的 ncRNA-eQTL。最后,我们开发了一个用户友好的数据库 ncRNA-eQTL(http://ibi.hzau.edu.cn/ncRNA-eQTL),用于免费搜索、浏览和下载所有的 ncRNA-eQTL。我们预计,这种综合全面的资源将有助于我们理解人类复杂表型变异的机制基础,特别是对于 ncRNA 和癌症相关的研究。