Department of Biology, Duke University, Durham, NC, 27708, USA.
Duke Lemur Center, Duke University, Durham, NC, 27705, USA.
Heredity (Edinb). 2020 Jan;124(1):236-251. doi: 10.1038/s41437-019-0260-9. Epub 2019 Aug 21.
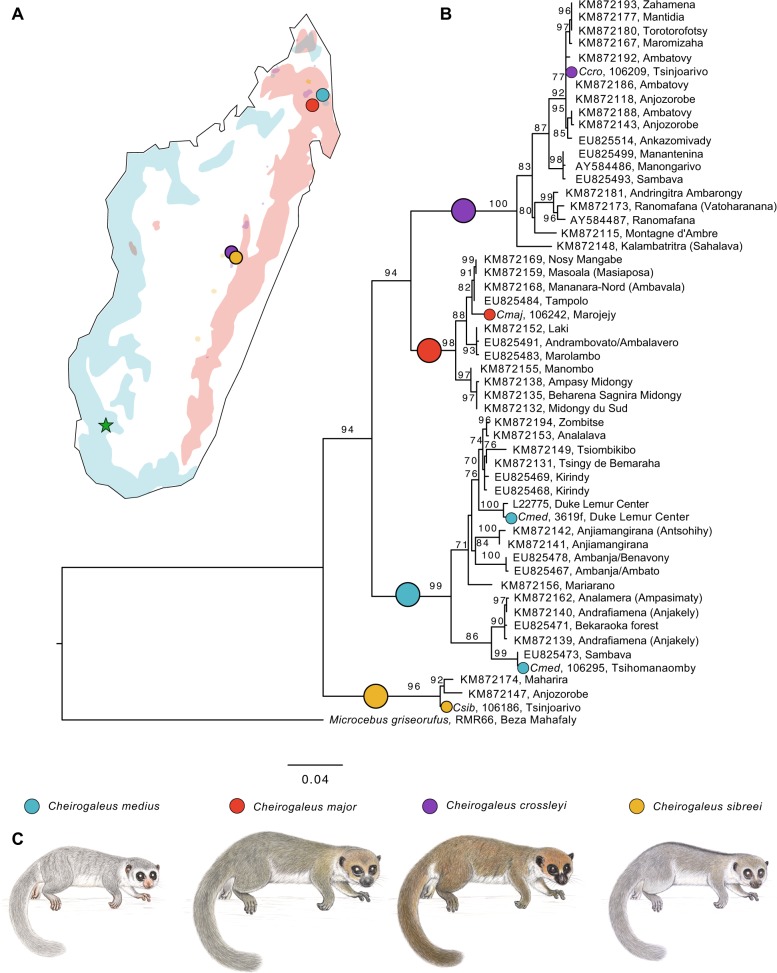
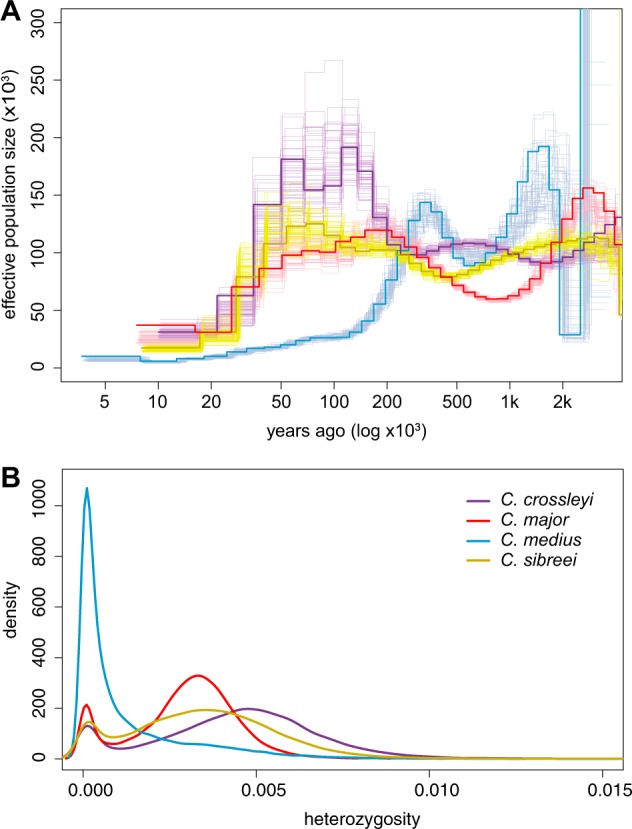
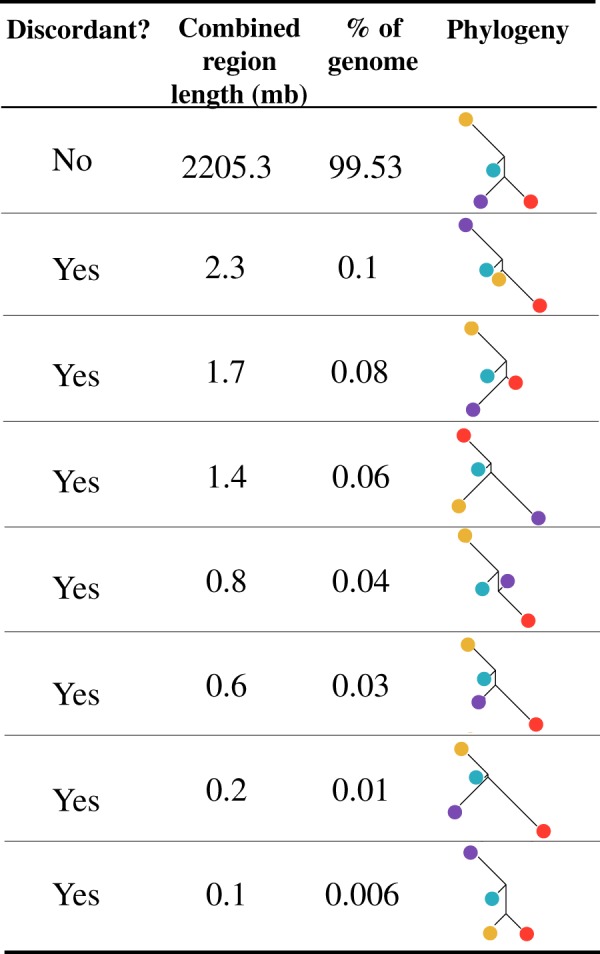
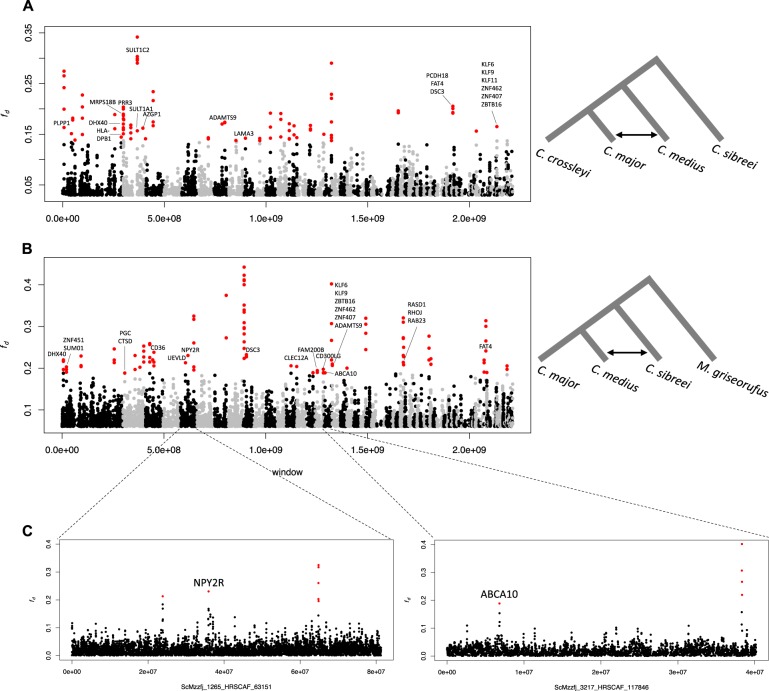
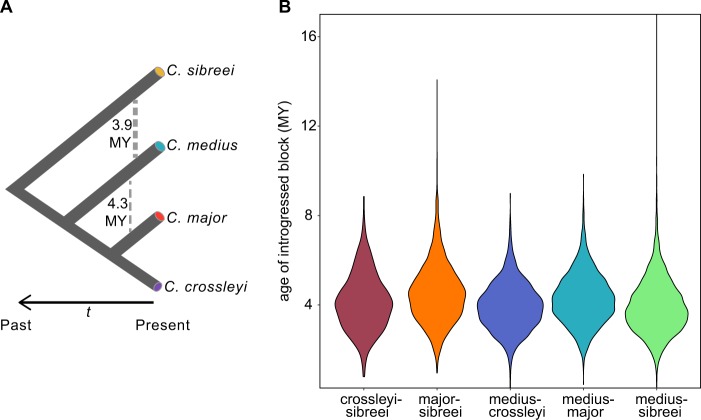
Madagascar's biodiversity is notoriously threatened by deforestation and climate change. Many of these organisms are rare, cryptic, and severely threatened, making population-level sampling unrealistic. Such is the case with Madagascar's dwarf lemurs (genus Cheirogaleus), the only obligate hibernating primate. We here apply comparative genomic approaches to generate the first genome-wide estimates of genetic diversity within dwarf lemurs. We generate a reference genome for the fat-tailed dwarf lemur, Cheirogaleus medius, and use this resource to facilitate analyses of high-coverage (~30×) genome sequences for wild-caught individuals representing species: C. sp. cf. medius, C. major, C. crossleyi, and C. sibreei. This study represents the largest contribution to date of novel genomic resources for Madagascar's lemurs. We find concordant phylogenetic relationships among the four lineages of Cheirogaleus across most of the genome, and yet detect a number of discordant genomic regions consistent with ancient admixture. We hypothesized that these regions could have resulted from adaptive introgression related to hibernation, indeed finding that genes associated with hibernation are present, though most significantly, that gene ontology categories relating to transcription are over-represented. We estimate levels of heterozygosity and find particularly low levels in an individual sampled from an isolated population of C. medius that we refer to as C. sp. cf. medius. Results are consistent with a recent decline in effective population size, which is evident across species. Our study highlights the power of comparative genomic analysis for identifying species and populations of conservation concern, as well as for illuminating possible mechanisms of adaptive phenotypic evolution.
马达加斯加的生物多样性因森林砍伐和气候变化而受到严重威胁。这些生物中有许多是稀有、隐蔽和严重受威胁的,因此无法进行种群水平的采样。马达加斯加的侏儒狐猴(Cheirogaleus 属)就是这种情况,它们是唯一的强制性冬眠灵长类动物。在这里,我们应用比较基因组学方法来生成侏儒狐猴内遗传多样性的第一个全基因组估计。我们为胖尾侏儒狐猴(Cheirogaleus medius)生成了一个参考基因组,并利用这一资源来促进对代表物种的野生捕获个体进行高覆盖率(约 30×)基因组序列的分析:C. sp. cf. medius、C. major、C. crossleyi 和 C. sibreei。这项研究代表了迄今为止对马达加斯加狐猴的新基因组资源的最大贡献。我们在 Cheirogaleus 的四个谱系中发现了大多数基因组上的一致的系统发育关系,但也检测到了一些不一致的基因组区域,这与古老的混合相一致。我们假设这些区域可能是与冬眠相关的适应性渗入的结果,实际上我们发现与冬眠相关的基因存在,尽管最显著的是,与转录相关的基因本体论类别过度表达。我们估计了杂合度水平,在我们称为 C. sp. cf. medius 的来自孤立的 C. medius 种群的个体中发现了特别低的水平。结果与有效种群大小的最近下降一致,这在各个物种中都很明显。我们的研究强调了比较基因组分析在识别保护关注的物种和种群以及阐明可能的适应性表型进化机制方面的强大功能。