Center for Molecular Biology of RNA, Department of Molecular, Cell & Developmental Biology, University of California, Santa Cruz, Santa Cruz, California, United States of America.
Department of Biomolecular Engineering, University of California, Santa Cruz, Santa Cruz, California, United States of America.
PLoS Genet. 2019 Aug 22;15(8):e1008249. doi: 10.1371/journal.pgen.1008249. eCollection 2019 Aug.
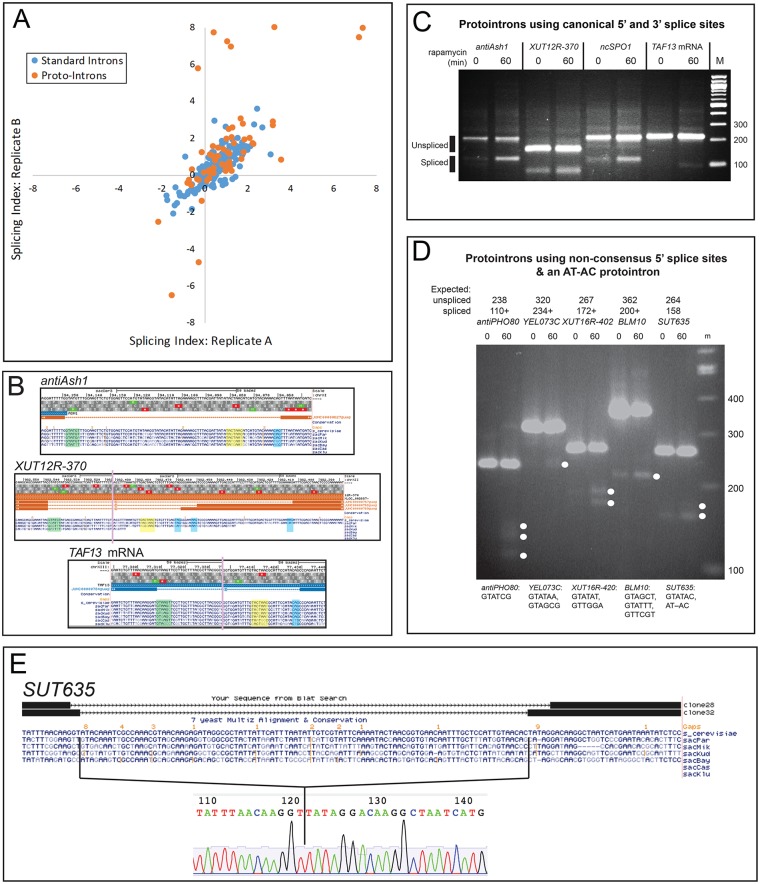
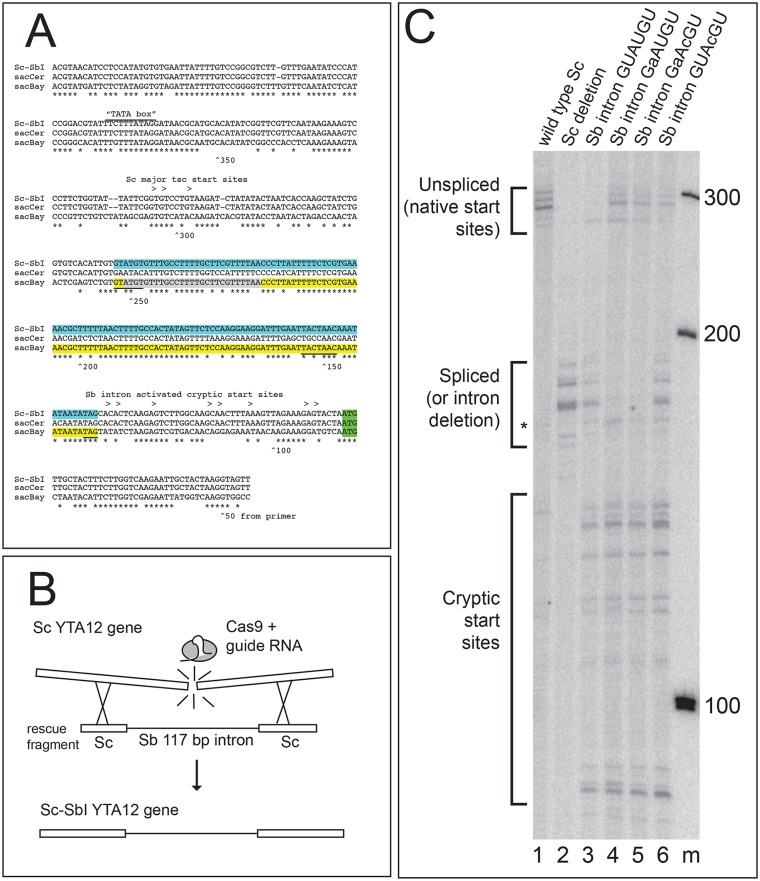
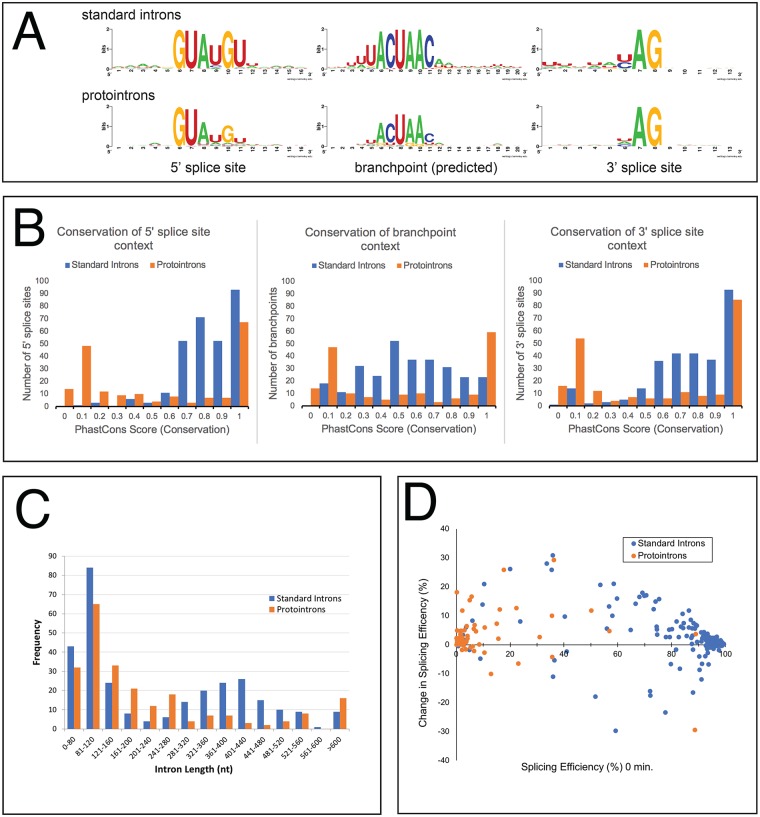
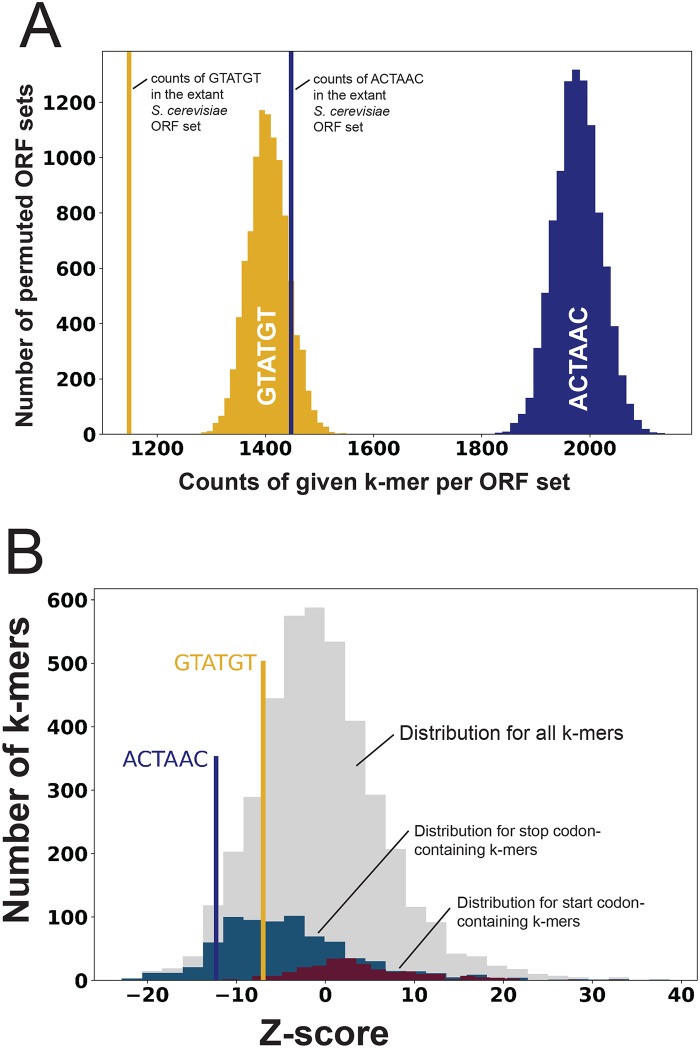
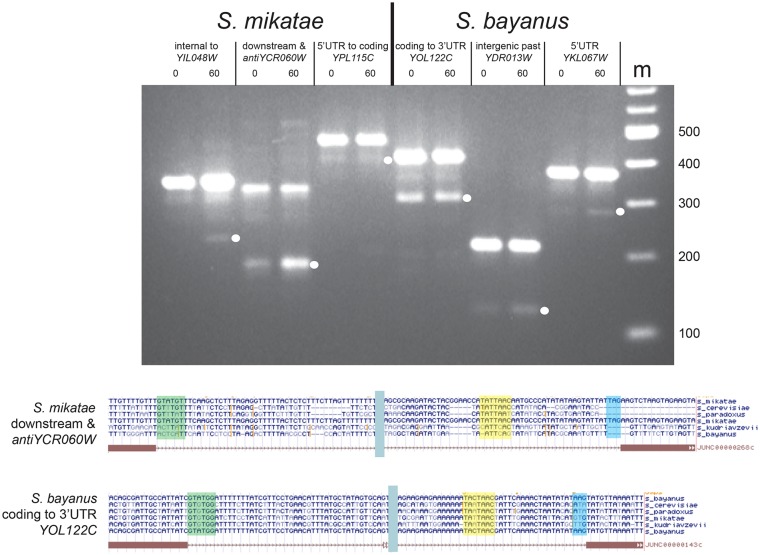
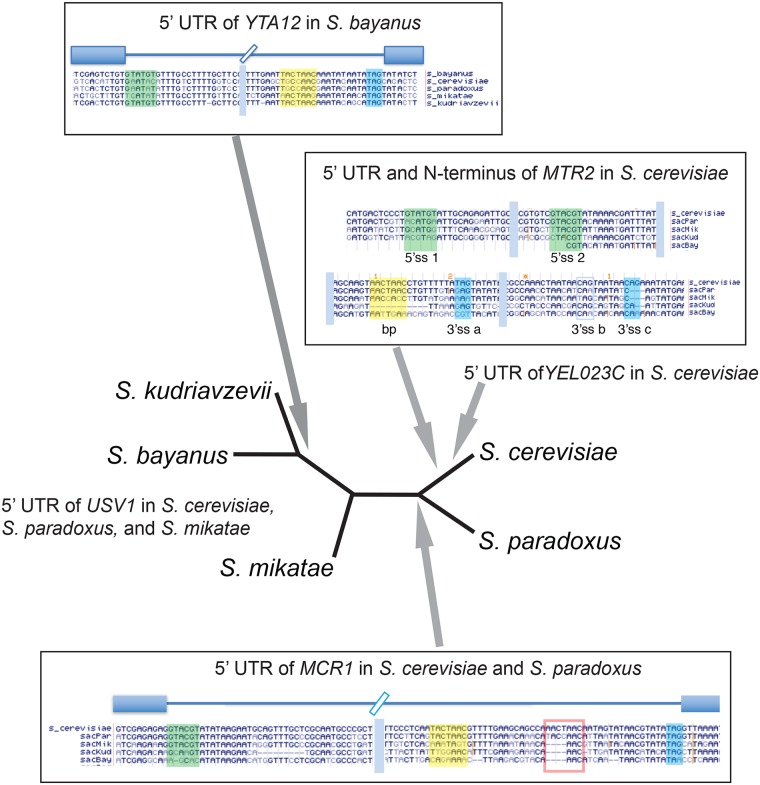
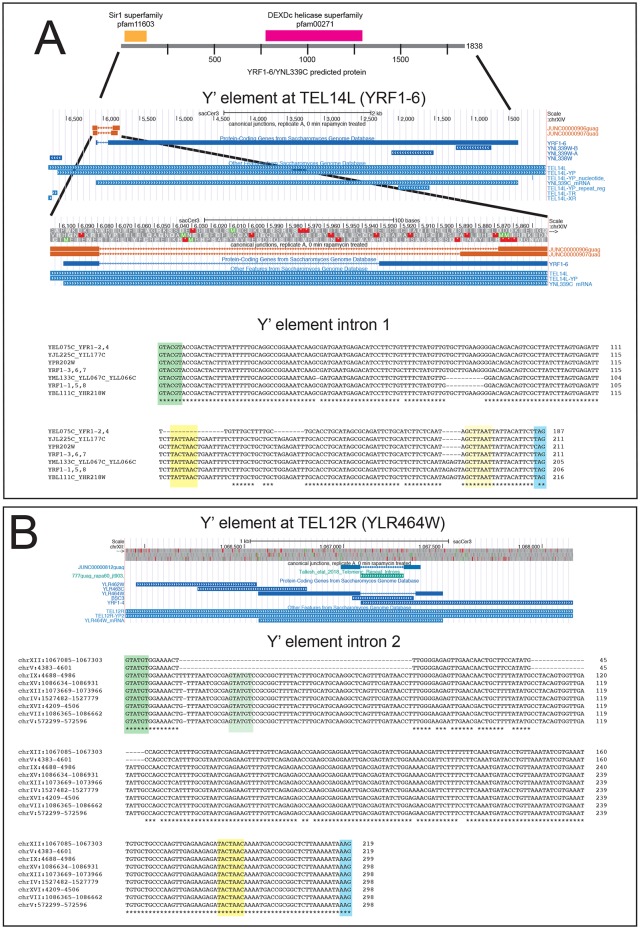
Introns are a prevalent feature of eukaryotic genomes, yet their origins and contributions to genome function and evolution remain mysterious. In budding yeast, repression of the highly transcribed intron-containing ribosomal protein genes (RPGs) globally increases splicing of non-RPG transcripts through reduced competition for the spliceosome. We show that under these "hungry spliceosome" conditions, splicing occurs at more than 150 previously unannotated locations we call protointrons that do not overlap known introns. Protointrons use a less constrained set of splice sites and branchpoints than standard introns, including in one case AT-AC in place of GT-AG. Protointrons are not conserved in all closely related species, suggesting that most are not under positive selection and are fated to disappear. Some are found in non-coding RNAs (e. g. CUTs and SUTs), where they may contribute to the creation of new genes. Others are found across boundaries between noncoding and coding sequences, or within coding sequences, where they offer pathways to the creation of new protein variants, or new regulatory controls for existing genes. We define protointrons as (1) nonconserved intron-like sequences that are (2) infrequently spliced, and importantly (3) are not currently understood to contribute to gene expression or regulation in the way that standard introns function. A very few protointrons in S. cerevisiae challenge this classification by their increased splicing frequency and potential function, consistent with the proposed evolutionary process of "intronization", whereby new standard introns are created. This snapshot of intron evolution highlights the important role of the spliceosome in the expansion of transcribed genomic sequence space, providing a pathway for the rare events that may lead to the birth of new eukaryotic genes and the refinement of existing gene function.
内含子是真核基因组的普遍特征,但它们的起源及其对基因组功能和进化的贡献仍然神秘。在出芽酵母中,通过减少对剪接体的竞争,高度转录的内含子核糖体蛋白基因(RPGs)的抑制会全局增加非-RPG 转录物的剪接。我们表明,在这些“饥饿剪接体”条件下,剪接发生在 150 多个以前未注释的位置,我们称之为原内含子,它们不与已知内含子重叠。原内含子使用比标准内含子更受限制的剪接位点和分支点集,包括在一种情况下,用 AT-AC 代替 GT-AG。原内含子在所有密切相关的物种中都没有保守,这表明大多数原内含子不受正选择的影响,注定会消失。有些存在于非编码 RNA(例如 CUTs 和 SUTs)中,它们可能有助于新基因的产生。其他存在于非编码和编码序列之间的边界处,或者在编码序列内,它们为产生新的蛋白质变体或现有基因的新调控提供了途径。我们将原内含子定义为(1)非保守的内含子样序列,(2)很少被剪接,重要的是(3)目前不被认为以与标准内含子功能相同的方式有助于基因表达或调控。少数原内含子在酿酒酵母中的高剪接频率和潜在功能挑战了这一分类,这与新的标准内含子的“内含子化”进化过程一致。这种内含子进化的快照突出了剪接体在转录基因组序列空间扩展中的重要作用,为可能导致新真核基因诞生和现有基因功能细化的罕见事件提供了途径。