Institute for Diabetes, Obesity, and Metabolism, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
Institute for Diabetes, Obesity, and Metabolism, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA; Department of Physiology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA.
Mol Metab. 2019 Oct;28:1-13. doi: 10.1016/j.molmet.2019.08.001. Epub 2019 Aug 5.
Skeletal muscle insulin signaling is a major determinant of muscle growth and glucose homeostasis. Protein kinase B/Akt plays a prominent role in mediating many of the metabolic effects of insulin. Mice and humans harboring systemic loss-of-function mutations in Akt2, the most abundant Akt isoform in metabolic tissues, are glucose intolerant and insulin resistant. Since the skeletal muscle accounts for a significant amount of postprandial glucose disposal, a popular hypothesis in the diabetes field suggests that a reduction in Akt, specifically in skeletal muscle, leads to systemic glucose intolerance and insulin resistance. Despite this common belief, the specific role of skeletal muscle Akt in muscle growth and insulin sensitivity remains undefined.
We generated multiple mouse models of skeletal muscle Akt deficiency to evaluate the role of muscle Akt signaling in vivo. The effects of these genetic perturbations on muscle mass, glucose homeostasis and insulin sensitivity were assessed using both in vivo and ex vivo assays.
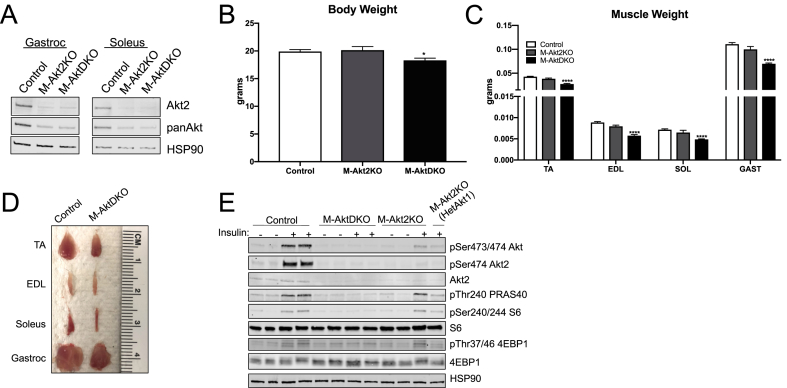
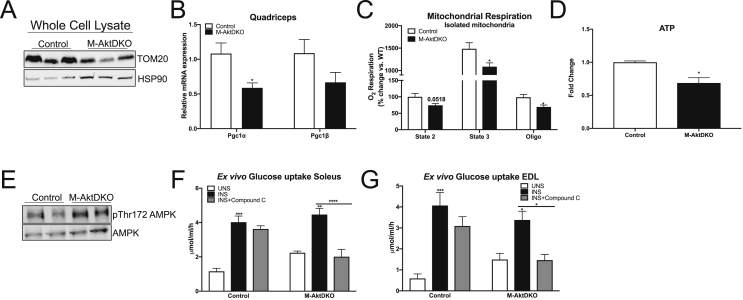
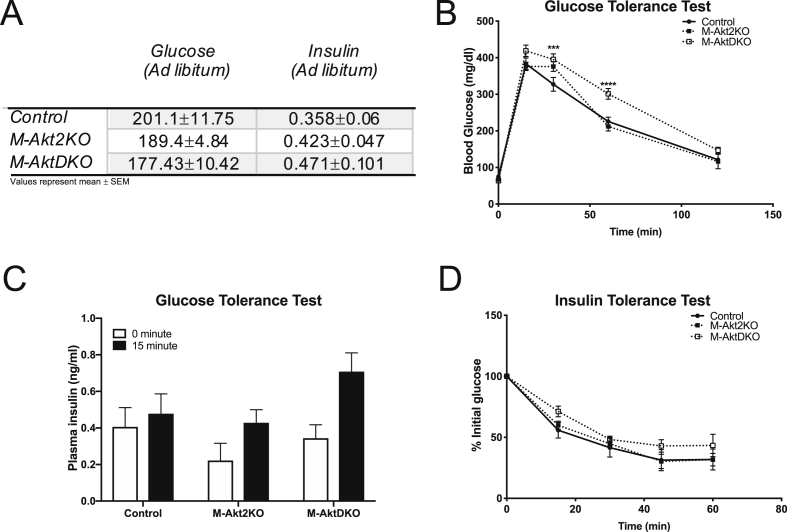
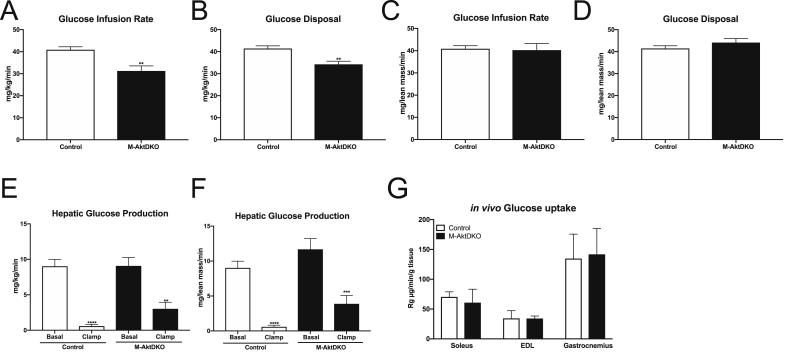
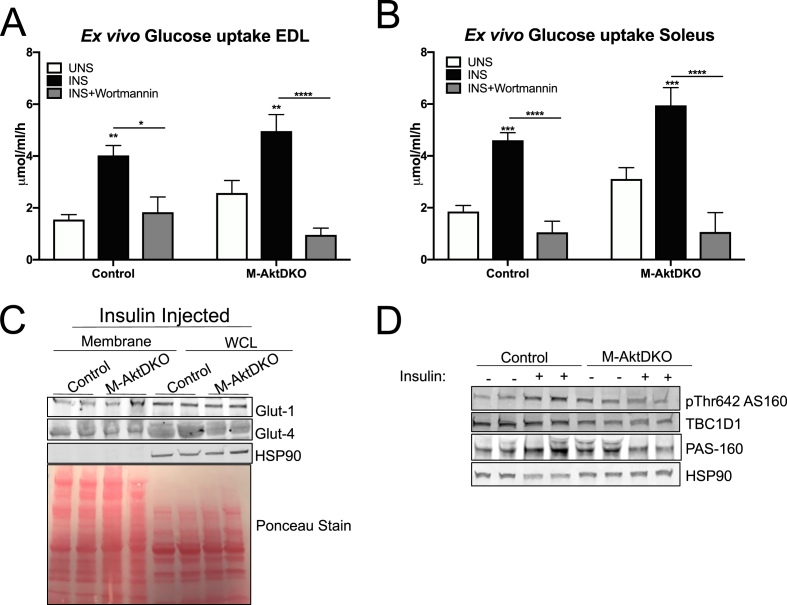
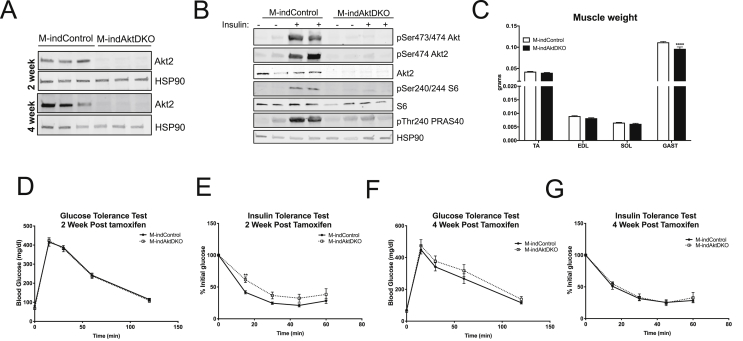
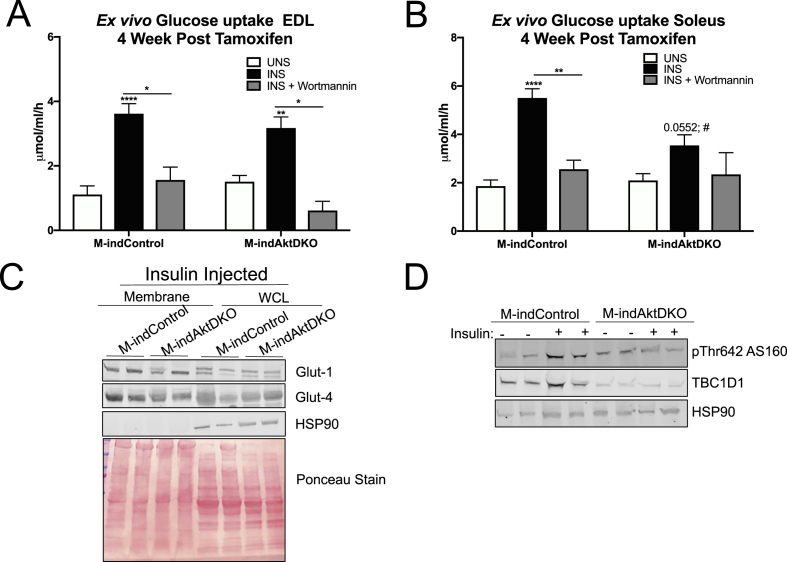
Surprisingly, mice lacking Akt2 alone in skeletal muscle displayed normal skeletal muscle insulin signaling, glucose tolerance, and insulin sensitivity despite a dramatic reduction in phosphorylated Akt. In contrast, deletion of both Akt isoforms (M-AktDKO) prevented downstream signaling and resulted in muscle atrophy. Despite the absence of Akt signaling, in vivo and ex vivo insulin-stimulated glucose uptake were normal in M-AktDKO mice. Similar effects on insulin sensitivity were observed in mice with prolonged deletion (4 weeks) of both skeletal muscle Akt isoforms selectively in adulthood. Conversely, short term deletion (2 weeks) of skeletal muscle specific Akt in adult muscles impaired insulin tolerance paralleling the effect observed by acute pharmacological inhibition of Akt in vitro. Mechanistically, chronic ablation of Akt induced mitochondrial dysfunction and activation of AMPK, which was required for insulin-stimulated glucose uptake in the absence of Akt.
Together, these data indicate that chronic reduction in Akt activity alone in skeletal muscle is not sufficient to induce insulin resistance or prevent glucose uptake in all conditions. Therefore, since insulin-stimulated glucose disposal in skeletal muscle is markedly impaired in insulin-resistant states, we hypothesize that alterations in signaling molecules in addition to skeletal muscle Akt are necessary to perturb glucose tolerance and insulin sensitivity in vivo.
骨骼肌胰岛素信号是肌肉生长和葡萄糖内稳态的主要决定因素。蛋白激酶 B/Akt 在介导胰岛素的许多代谢作用中起着突出的作用。在代谢组织中最丰富的 Akt 同工型 Akt2 中全身丧失功能的突变的小鼠和人类是葡萄糖不耐受和胰岛素抵抗的。由于骨骼肌占餐后葡萄糖处置的很大一部分,糖尿病领域的一个流行假设是 Akt 的减少,特别是在骨骼肌中,导致全身葡萄糖不耐受和胰岛素抵抗。尽管存在这种普遍的信念,但骨骼肌 Akt 在肌肉生长和胰岛素敏感性中的具体作用仍未定义。
我们生成了多种骨骼肌 Akt 缺乏的小鼠模型,以评估肌肉 Akt 信号在体内的作用。使用体内和体外测定法评估这些遗传干扰对肌肉质量、葡萄糖稳态和胰岛素敏感性的影响。
令人惊讶的是,骨骼肌中单独缺乏 Akt2 的小鼠表现出正常的骨骼肌胰岛素信号、葡萄糖耐量和胰岛素敏感性,尽管磷酸化 Akt 显著减少。相比之下,两种 Akt 同工型(M-AktDKO)的缺失阻止了下游信号传导并导致肌肉萎缩。尽管缺乏 Akt 信号,但在 M-AktDKO 小鼠中,体内和体外胰岛素刺激的葡萄糖摄取均正常。在成年期选择性长时间(4 周)缺失两种骨骼肌 Akt 同工型的小鼠中也观察到类似的胰岛素敏感性影响。相反,在成年肌肉中短期(2 周)缺失骨骼肌特异性 Akt 会损害胰岛素耐量,这与体外急性药理学抑制 Akt 观察到的效果一致。从机制上讲,慢性 Akt 消融诱导线粒体功能障碍和 AMPK 激活,这对于缺乏 Akt 时的胰岛素刺激的葡萄糖摄取是必需的。
总的来说,这些数据表明,在骨骼肌中单独慢性降低 Akt 活性不足以在所有情况下诱导胰岛素抵抗或阻止葡萄糖摄取。因此,由于在胰岛素抵抗状态下骨骼肌中胰岛素刺激的葡萄糖摄取明显受损,我们假设除了骨骼肌 Akt 之外,信号分子的改变对于在体内扰乱葡萄糖耐量和胰岛素敏感性是必要的。