Guin Debleena, Rani Jyoti, Singh Priyanka, Grover Sandeep, Bora Shivangi, Talwar Puneet, Karthikeyan Muthusamy, Satyamoorthy K, Adithan C, Ramachandran S, Saso Luciano, Hasija Yasha, Kukreti Ritushree
Genomics and Molecular Medicine Unit, Council of Scientific and Industrial Research (CSIR)-Institute of Genomics and Integrative Biology (IGIB), New Delhi, India.
Department of Biotechnology, Delhi Technological University, Delhi, India.
Front Pharmacol. 2019 Aug 7;10:839. doi: 10.3389/fphar.2019.00839. eCollection 2019.
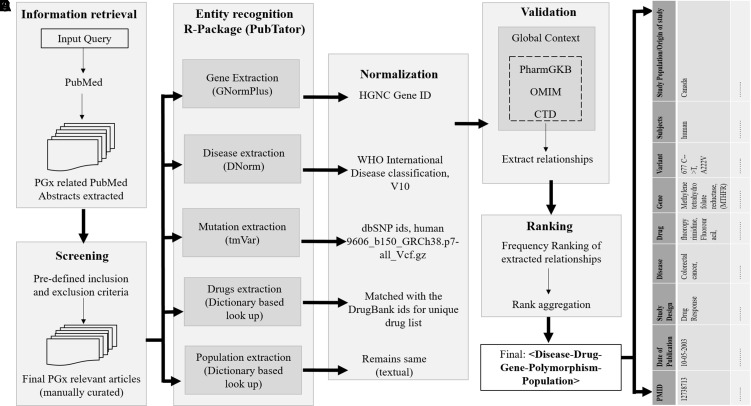
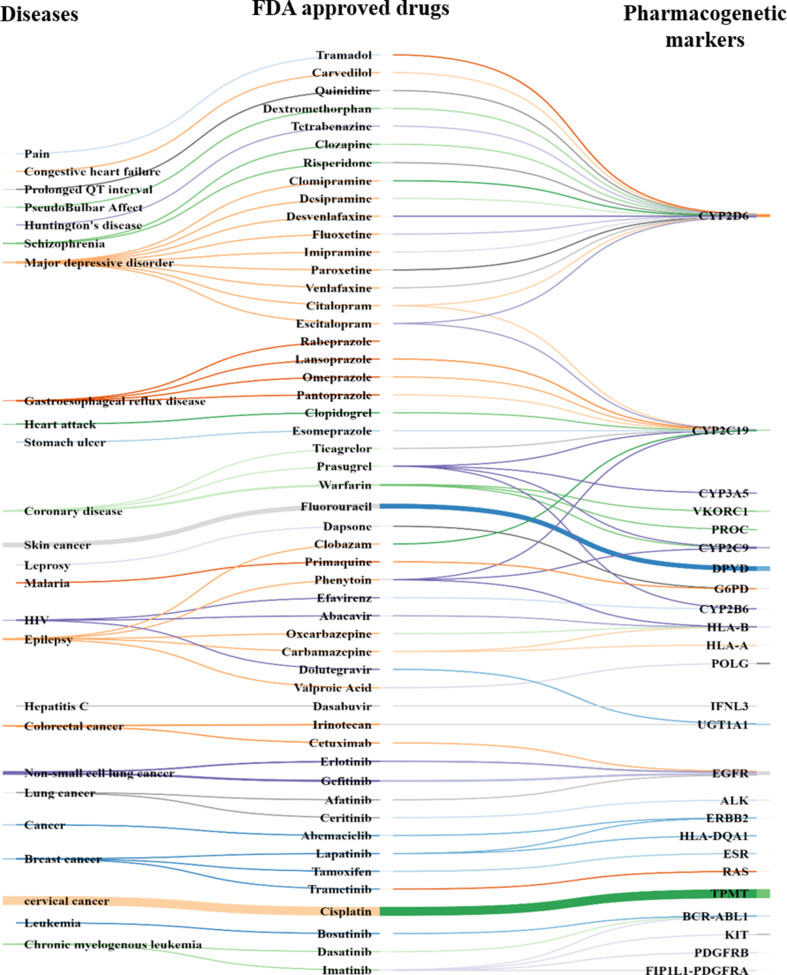
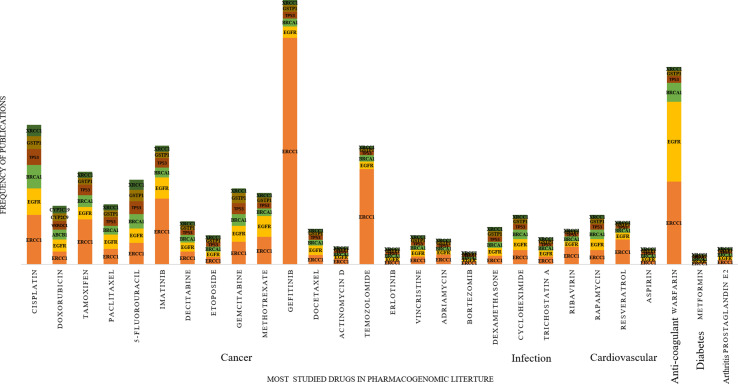
Understanding patients' genomic variations and their effect in protecting or predisposing them to drug response phenotypes is important for providing personalized healthcare. Several studies have manually curated such genotype-phenotype relationships into organized databases from clinical trial data or published literature. However, there are no text mining tools available to extract high-accuracy information from such existing knowledge. In this work, we used a semiautomated text mining approach to retrieve a complete pharmacogenomic (PGx) resource integrating disease-drug-gene-polymorphism relationships to derive a global perspective for ease in therapeutic approaches. We used an R package, pubmed.mineR, to automatically retrieve PGx-related literature. We identified 1,753 disease types, and 666 drugs, associated with 4,132 genes and 33,942 polymorphisms collated from 180,088 publications. With further manual curation, we obtained a total of 2,304 PGx relationships. We evaluated our approach by performance (precision = 0.806) with benchmark datasets like Pharmacogenomic Knowledgebase (PharmGKB) (0.904), Online Mendelian Inheritance in Man (OMIM) (0.600), and The Comparative Toxicogenomics Database (CTD) (0.729). We validated our study by comparing our results with 362 commercially used the US- Food and drug administration (FDA)-approved drug labeling biomarkers. Of the 2,304 PGx relationships identified, 127 belonged to the FDA list of 362 approved pharmacogenomic markers, indicating that our semiautomated text mining approach may reveal significant PGx information with markers for drug response prediction. In addition, it is a scalable and state-of-art approach in curation for PGx clinical utility.
了解患者的基因组变异及其在保护或使他们易患药物反应表型方面的作用,对于提供个性化医疗保健至关重要。几项研究已从临床试验数据或已发表的文献中手动整理了此类基因型-表型关系,并将其纳入有组织的数据库中。然而,目前还没有文本挖掘工具可用于从此类现有知识中提取高精度信息。在这项工作中,我们使用了一种半自动文本挖掘方法来检索一个完整的药物基因组学(PGx)资源,该资源整合了疾病-药物-基因-多态性之间的关系,以便从全局角度轻松制定治疗方法。我们使用一个R包pubmed.mineR自动检索与PGx相关的文献。我们从180,088篇出版物中整理出1,753种疾病类型、666种药物,这些与4,132个基因和33,942个多态性相关。经过进一步的人工整理,我们总共获得了2,304个PGx关系。我们使用诸如药物基因组学知识库(PharmGKB)(精度 = 0.904)、《人类孟德尔遗传在线》(OMIM)(精度 = 0.600)和比较毒理基因组学数据库(CTD)(精度 = 0.729)等基准数据集来评估我们方法的性能(精度 = 0.806)。我们通过将我们的结果与362种商业使用的美国食品药品监督管理局(FDA)批准的药物标签生物标志物进行比较来验证我们的研究。在确定的2,304个PGx关系中,有127个属于FDA列出的362个批准的药物基因组学标记,这表明我们的半自动文本挖掘方法可能通过药物反应预测标记揭示重要的PGx信息。此外,它是一种用于PGx临床应用整理的可扩展且先进的方法。