School of Informatics, Xiamen University, Xiamen 361005, China.
Graduate School, Yunnan Minzu University, Kunming 650504, China.
Cells. 2019 Aug 26;8(9):977. doi: 10.3390/cells8090977.
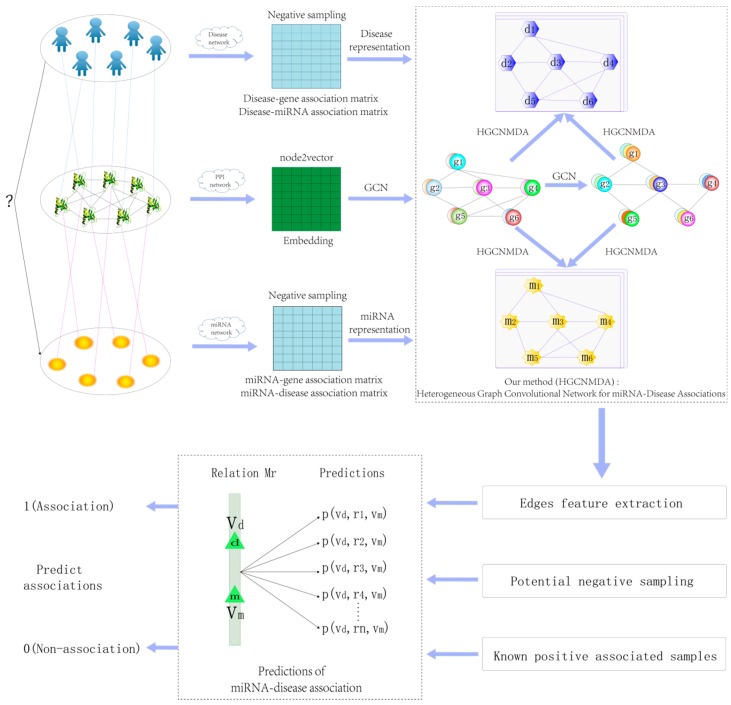
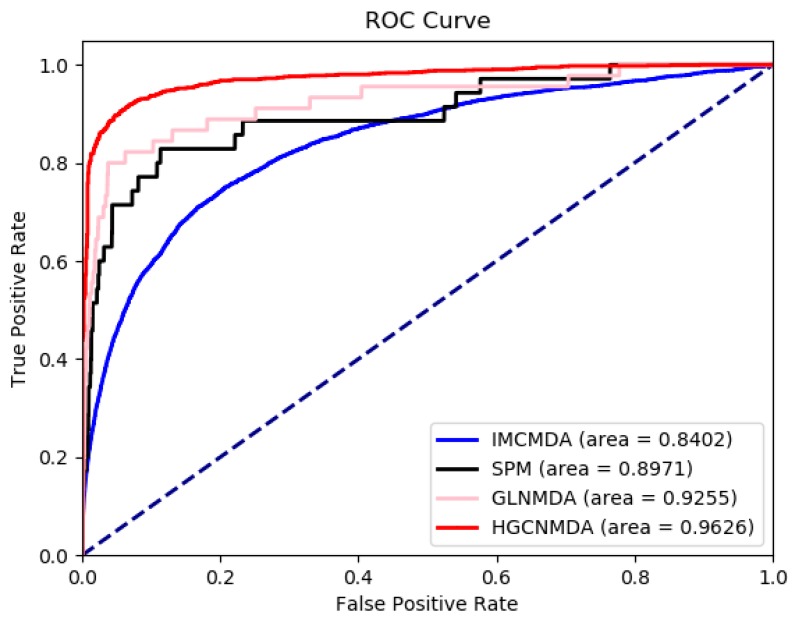
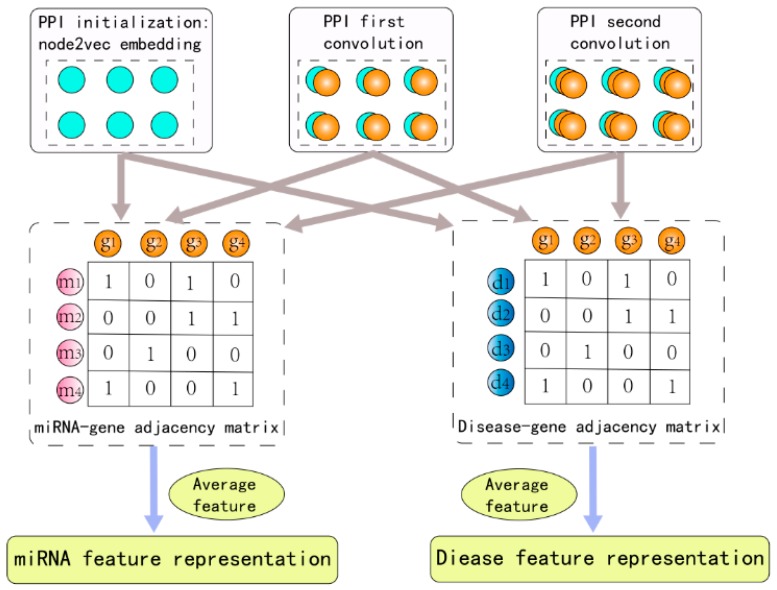
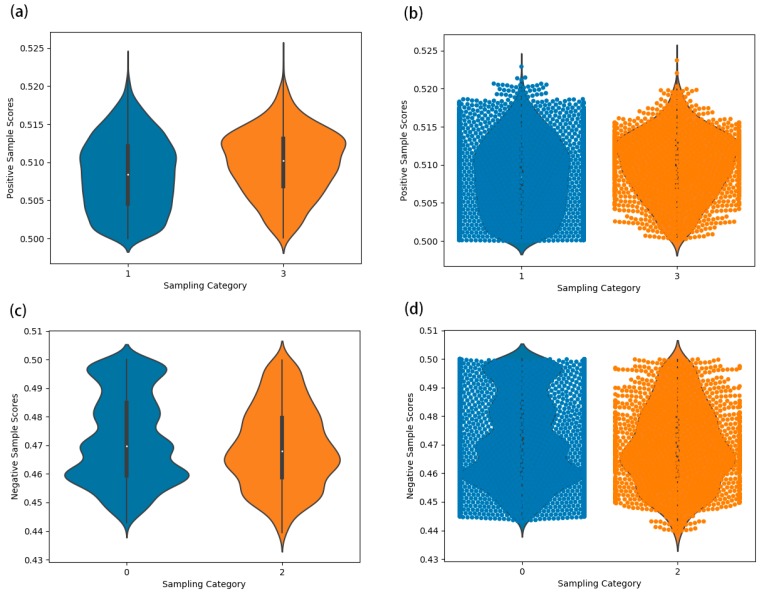
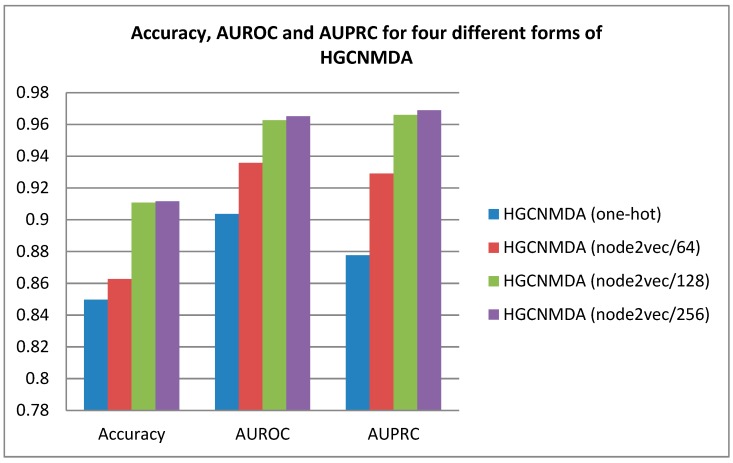
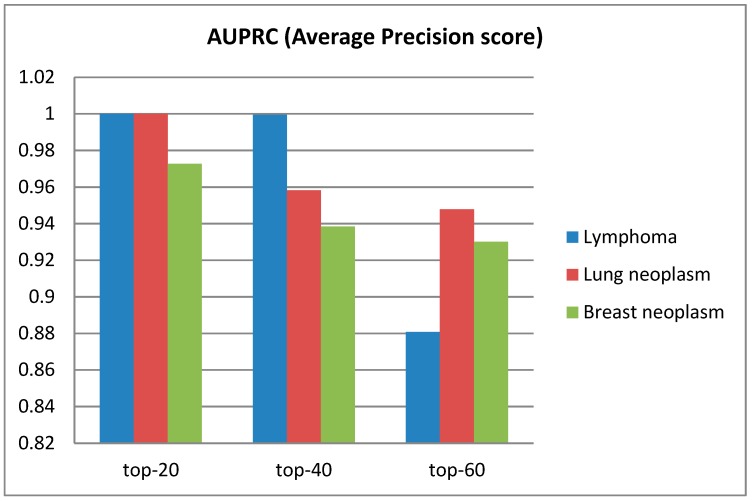
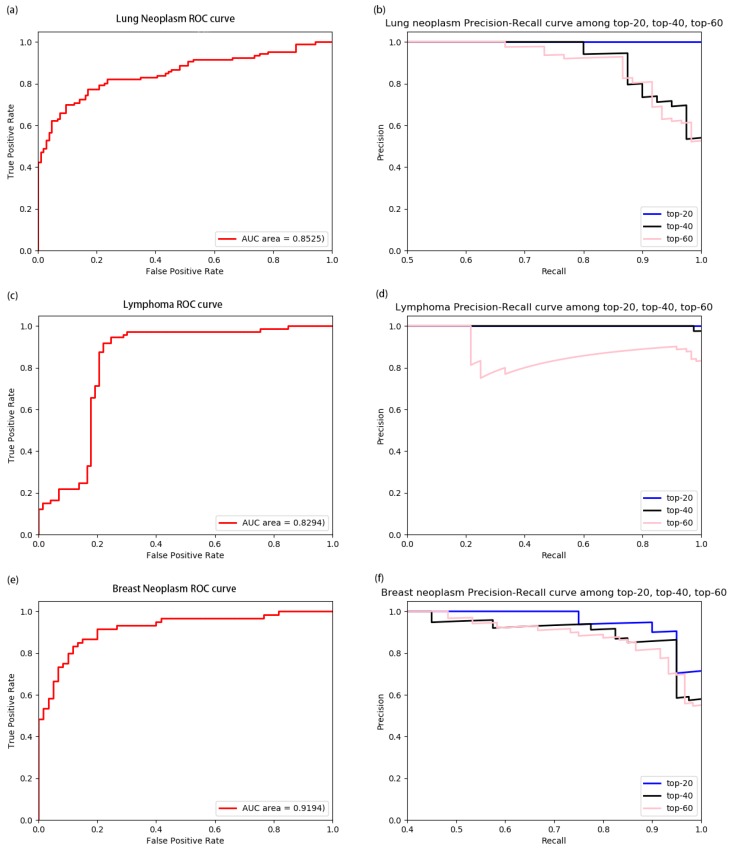
Identifying the interactions between disease and microRNA (miRNA) can accelerate drugs development, individualized diagnosis, and treatment for various human diseases. However, experimental methods are time-consuming and costly. So computational approaches to predict latent miRNA-disease interactions are eliciting increased attention. But most previous studies have mainly focused on designing complicated similarity-based methods to predict latent interactions between miRNAs and diseases. In this study, we propose a novel computational model, termed heterogeneous graph convolutional network for miRNA-disease associations (HGCNMDA), which is based on known human protein-protein interaction (PPI) and integrates four biological networks: miRNA-disease, miRNA-gene, disease-gene, and PPI network. HGCNMDA achieved reliable performance using leave-one-out cross-validation (LOOCV). HGCNMDA is then compared to three state-of-the-art algorithms based on five-fold cross-validation. HGCNMDA achieves an AUC of 0.9626 and an average precision of 0.9660, respectively, which is ahead of other competitive algorithms. We further analyze the top-10 unknown interactions between miRNA and disease. In summary, HGCNMDA is a useful computational model for predicting miRNA-disease interactions.
鉴定疾病与 microRNA(miRNA)之间的相互作用可以加速各种人类疾病的药物开发、个性化诊断和治疗。然而,实验方法既耗时又昂贵。因此,预测潜在 miRNA-疾病相互作用的计算方法引起了越来越多的关注。但之前的大多数研究主要集中在设计复杂的基于相似性的方法来预测 miRNA 和疾病之间的潜在相互作用。在这项研究中,我们提出了一种新的计算模型,称为基于异构图卷积网络的 miRNA-疾病关联模型(HGCNMDA),它基于已知的人类蛋白质-蛋白质相互作用(PPI),并整合了四个生物网络:miRNA-疾病、miRNA-基因、疾病-基因和 PPI 网络。HGCNMDA 使用留一法交叉验证(LOOCV)实现了可靠的性能。HGCNMDA 然后与基于五折交叉验证的三种最先进的算法进行比较。HGCNMDA 的 AUC 为 0.9626,平均精度为 0.9660,均优于其他竞争算法。我们进一步分析了 miRNA 和疾病之间的前 10 个未知相互作用。总之,HGCNMDA 是一种预测 miRNA-疾病相互作用的有用计算模型。