Division of Human Genetics, Children's Hospital of Philadelphia, Philadelphia, PA, USA.
Department of Pediatrics, Sapienza University of Rome, Rome, Italy.
Genet Med. 2020 Feb;22(2):326-335. doi: 10.1038/s41436-019-0645-4. Epub 2019 Sep 2.
The 22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion in humans, with highly variable phenotypic expression. Whereas congenital heart defects, palatal anomalies, immunodeficiency, hypoparathyroidism, and neuropsychiatric conditions are observed in over 50% of patients with 22q11DS, a subset of patients present with additional "atypical" findings such as craniosynostosis and anorectal malformations. Recently, pathogenic variants in the CDC45 (Cell Division Cycle protein 45) gene, located within the LCR22A-LCR22B region of chromosome 22q11.2, were noted to be involved in the pathogenesis of craniosynostosis.
We performed next-generation sequencing on DNA from 15 patients with 22q11.2DS and atypical phenotypic features such as craniosynostosis, short stature, skeletal differences, and anorectal malformations.
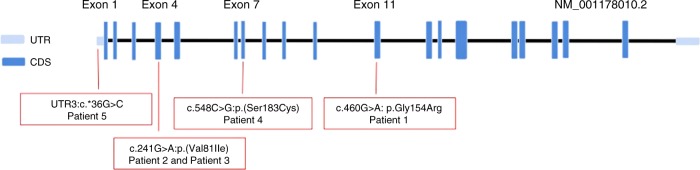
We identified four novel rare nonsynonymous variants in CDC45 in 5/15 patients with 22q11.2DS and craniosynostosis and/or other atypical findings.
This study supports CDC45 as a causative gene in craniosynostosis, as well as a number of other anomalies. We suggest that this association results in a condition independent of Meier-Gorlin syndrome, perhaps representing a novel condition and/or a cause of features associated with Baller-Gerold syndrome. In addition, this work confirms that the phenotypic variability observed in a subset of patients with 22q11.2DS is due to pathogenic variants on the nondeleted chromosome.
22q11.2 缺失综合征(22q11.2DS)是人类最常见的微缺失,具有高度可变的表型表达。虽然超过 50%的 22q11DS 患者存在先天性心脏病、腭畸形、免疫缺陷、甲状旁腺功能减退和神经精神疾病,但一部分患者还存在其他“非典型”表现,如颅缝早闭和肛门直肠畸形。最近,位于染色体 22q11.2 的 LCR22A-LCR22B 区域内的 CDC45(细胞分裂周期蛋白 45)基因的致病性变异被认为与颅缝早闭的发病机制有关。
我们对 15 名患有 22q11.2DS 和非典型表型特征(如颅缝早闭、身材矮小、骨骼差异和肛门直肠畸形)的患者的 DNA 进行了下一代测序。
我们在 5/15 名患有 22q11.2DS 伴颅缝早闭和/或其他非典型表现的患者中发现了 CDC45 的四个新的罕见非同义变异。
这项研究支持 CDC45 是颅缝早闭以及许多其他异常的致病基因。我们建议这种关联导致一种独立于 Meier-Gorlin 综合征的疾病,可能代表一种新的疾病或 Baller-Gerold 综合征相关特征的原因。此外,这项工作证实了在 22q11.2DS 的一部分患者中观察到的表型变异性是由于非缺失染色体上的致病性变异引起的。