KU Leuven, Department of Electrical Engineering (ESAT), STADIUS Center for Dynamical Systems, Signal Processing and Data Analytics, Kasteelpark Arenberg 10, Box 2446, 3001 Leuven, Belgium ; iMinds Medical IT Department, Kasteelpark Arenberg 10, Box 2446, 3001 Leuven, Belgium.
KU Leuven, Center of Human Genetics Gasthuisberg, O&N I Herestraat 49, Box 602, 3000 Leuven, Belgium.
Genome Med. 2014 Sep 17;6(9):71. doi: 10.1186/s13073-014-0071-9. eCollection 2014.
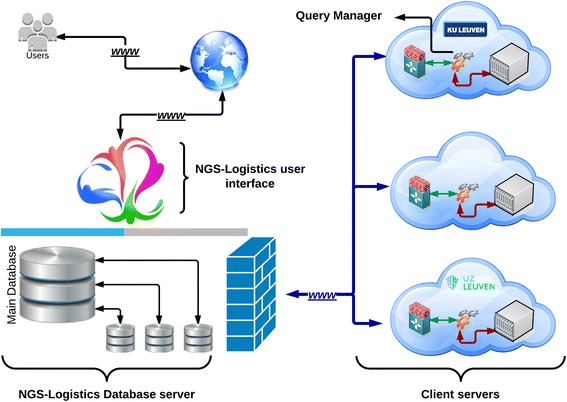
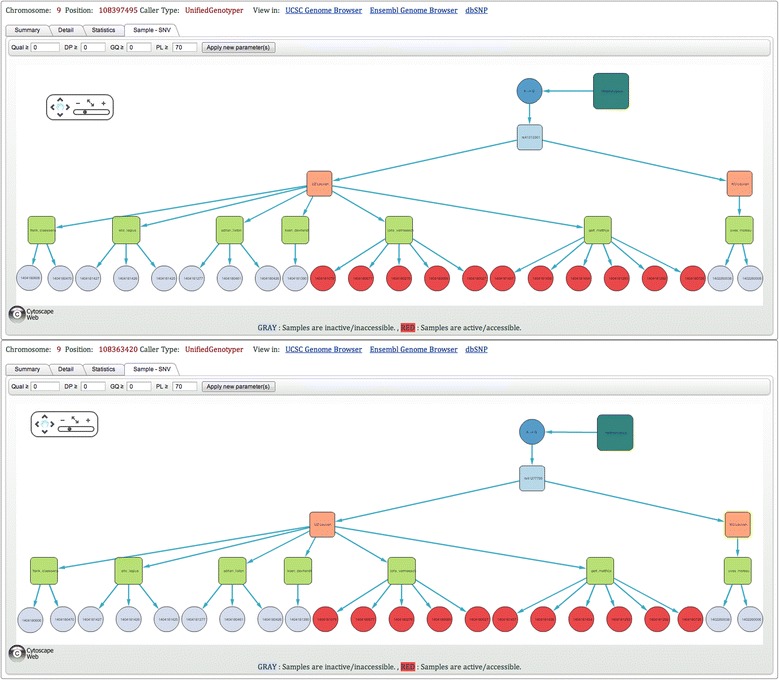
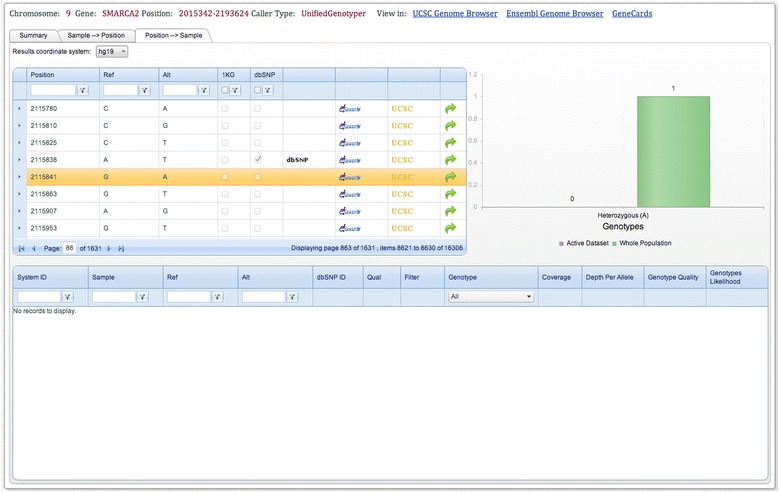
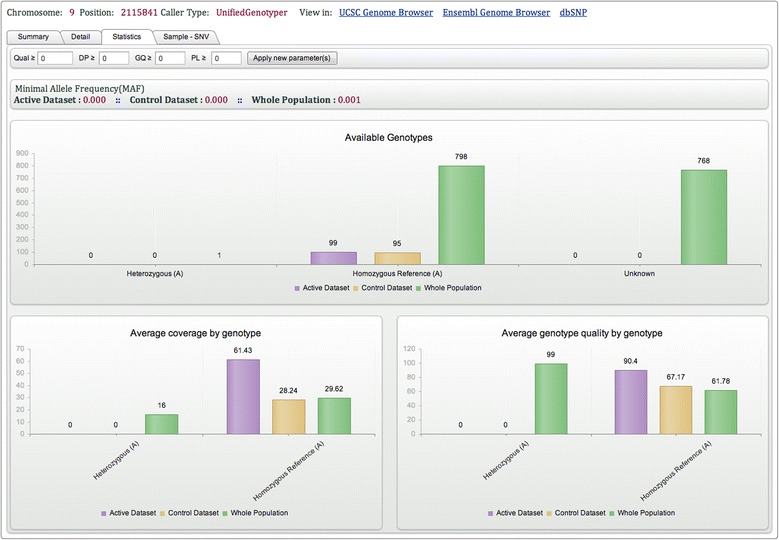
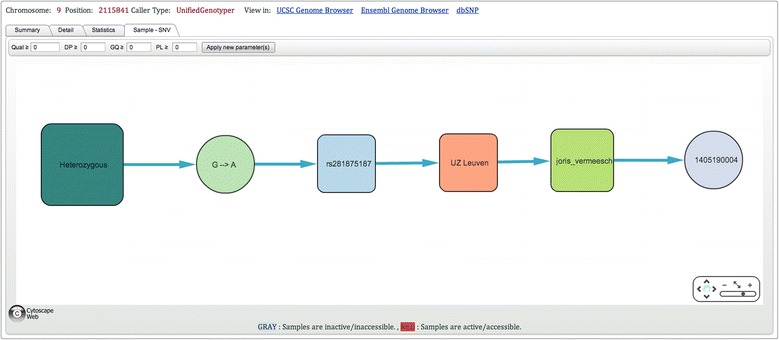
As many personal genomes are being sequenced, collaborative analysis of those genomes has become essential. However, analysis of personal genomic data raises important privacy and confidentiality issues. We propose a methodology for federated analysis of sequence variants from personal genomes. Specific base-pair positions and/or regions are queried for samples to which the user has access but also for the whole population. The statistics results do not breach data confidentiality but allow further exploration of the data; researchers can negotiate access to relevant samples through pseudonymous identifiers. This approach minimizes the impact on data confidentiality while enabling powerful data analysis by gaining access to important rare samples. Our methodology is implemented in an open source tool called NGS-Logistics, freely available at https://ngsl.esat.kuleuven.be.
随着越来越多的个人基因组被测序,对这些基因组进行协作分析已变得至关重要。然而,个人基因组数据的分析引发了重要的隐私和保密性问题。我们提出了一种用于对个人基因组序列变体进行联合分析的方法。针对用户可访问的样本以及整个群体,对特定的碱基对位置和/或区域进行查询。统计结果不会泄露数据机密性,但允许对数据进行进一步探索;研究人员可以通过匿名标识符协商访问相关样本。这种方法最大限度地减少了对数据保密性的影响,同时通过访问重要的稀有样本,实现了强大的数据分析。我们的方法在一个名为 NGS-Logistics 的开源工具中实现,可在 https://ngsl.esat.kuleuven.be 免费获得。