CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China.
College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China.
Front Immunol. 2019 Aug 16;10:1843. doi: 10.3389/fimmu.2019.01843. eCollection 2019.
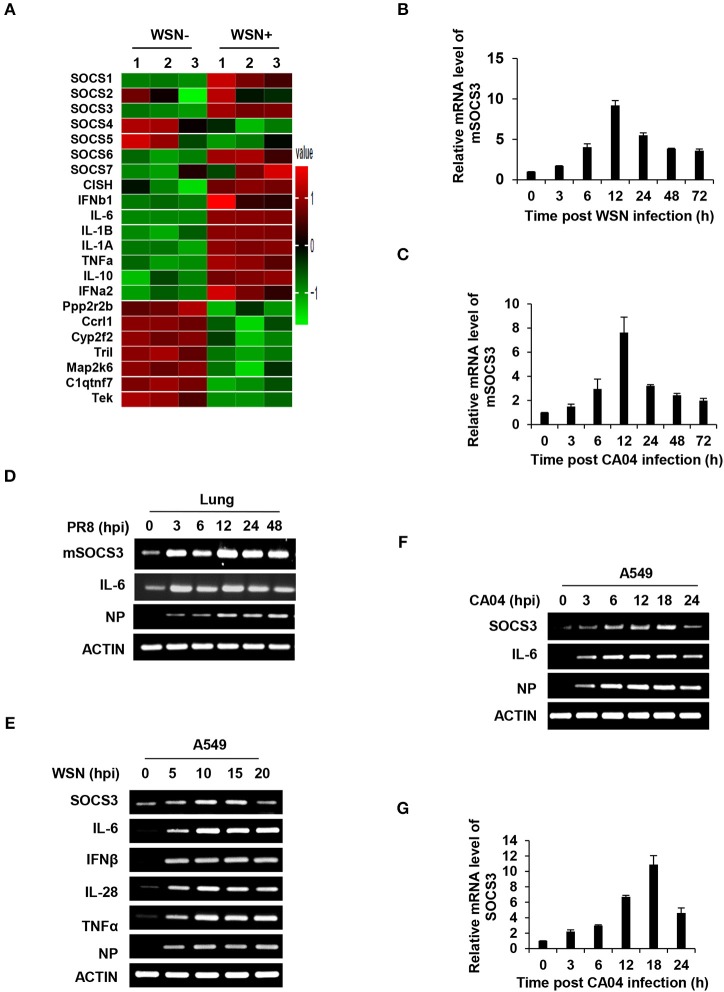
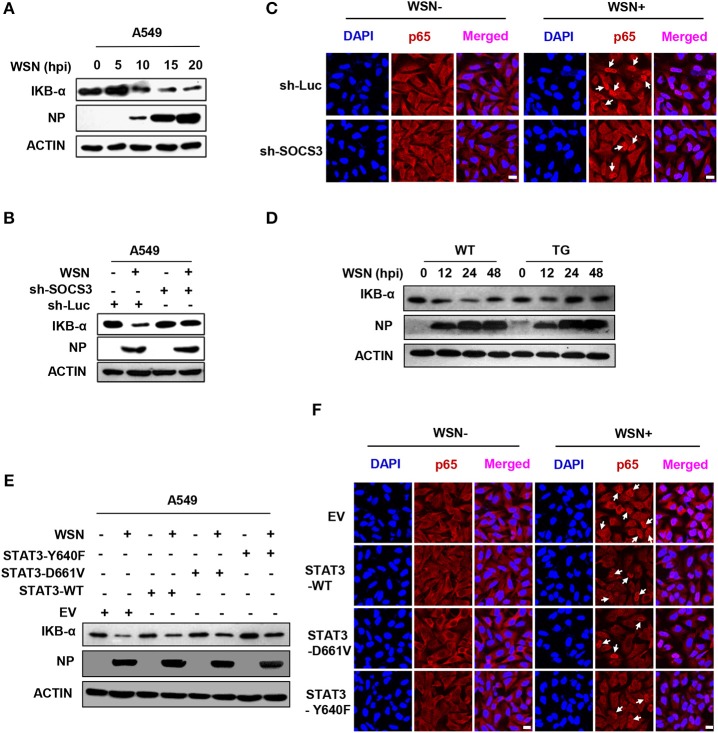
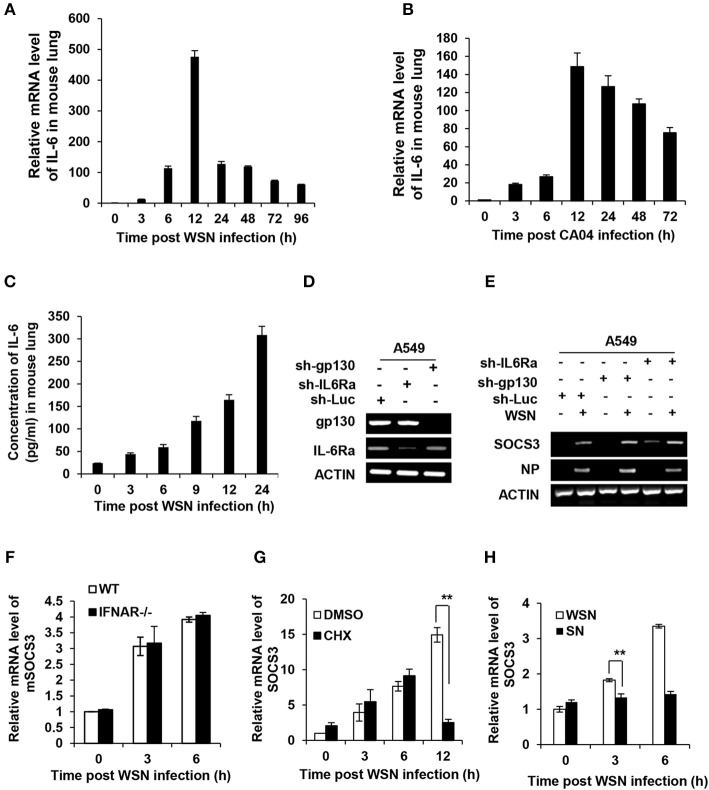
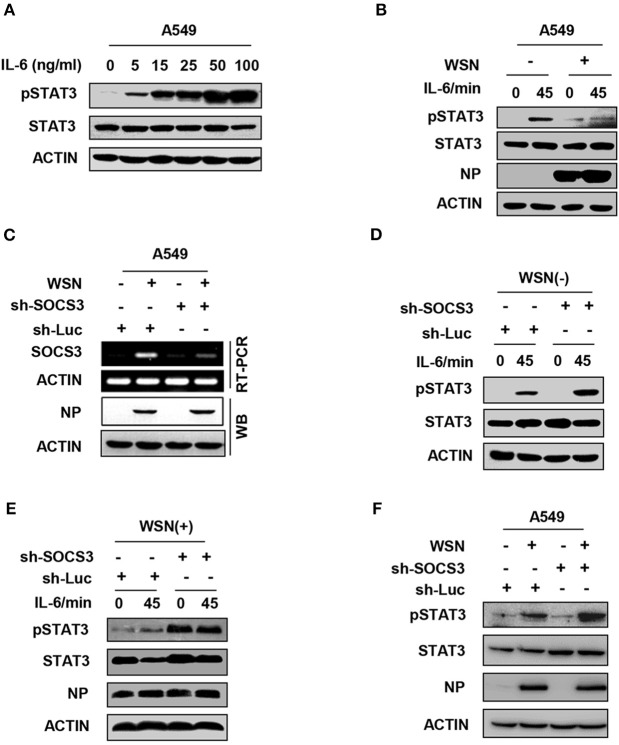
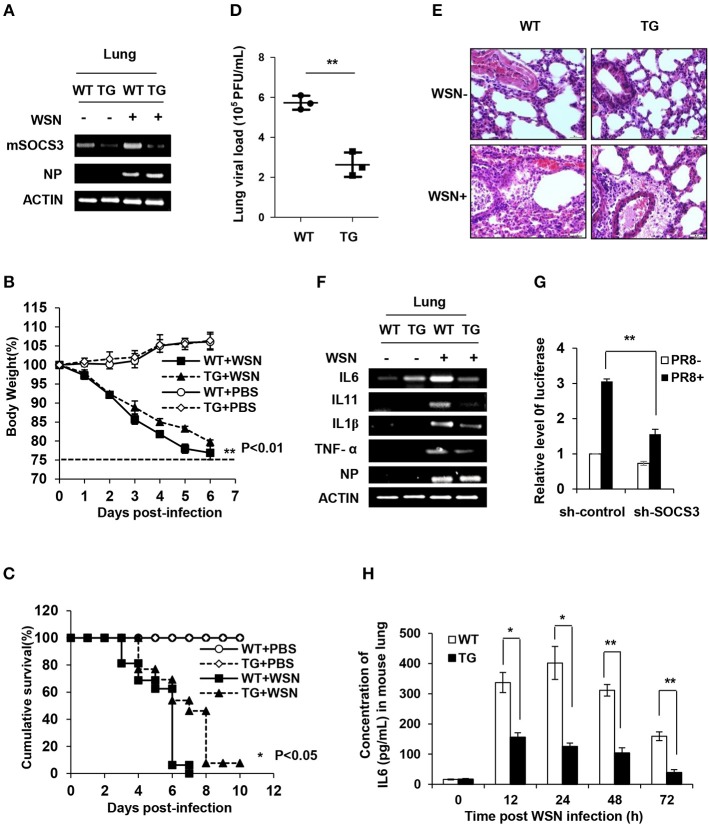
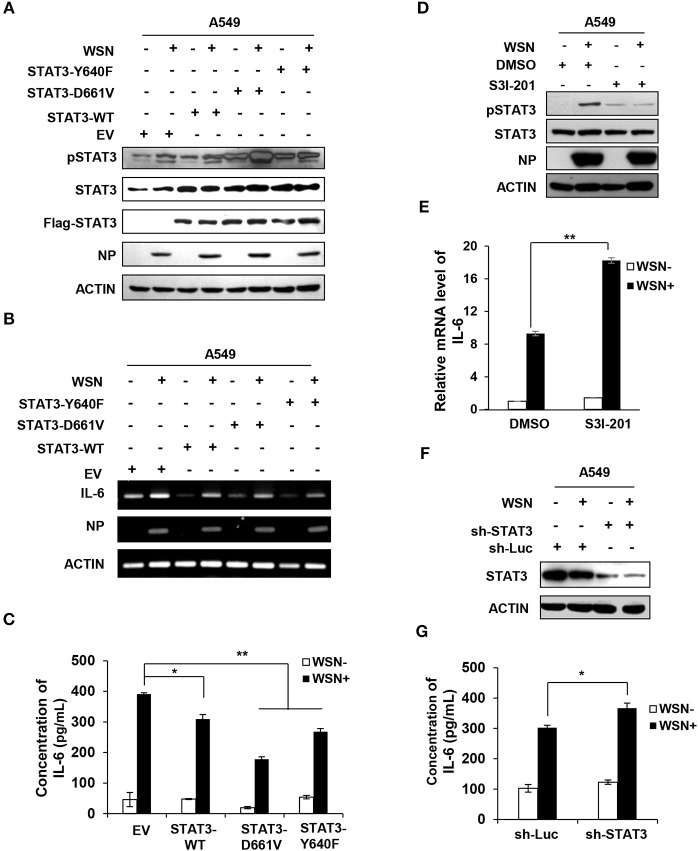
Influenza A virus (IAV) remains a major public health threat in the world, as indicated by the severe pneumonia caused by its infection annually. Interleukin-6 (IL-6) involved excessive inflammatory response to IAV infection profoundly contributes to the virus pathogenesis. However, the precise mechanisms underlying such a response are poorly understood. Here we found from both and studies that IAV not only induced a surge of IL-6 release, but also greatly upregulated expression of suppressor of cytokine signaling-3 (SOCS3), the potent suppressor of IL-6-associated signal transducer and activator of transcription 3 (STAT3) signaling. Interestingly, there existed a cytokine-independent mechanism of the robust induction of SOCS3 by IAV at early stages of the infection. Furthermore, we employed SOCS3-knockdown transgenic mice (TG), and surprisingly observed from virus challenge experiments using these mice that disruption of SOCS3 expression provided significant protection against IAV infection, as evidenced by attenuated acute lung injury, a higher survival rate of infected animals and lower viral load in infected tissues as compared with those of wild-type littermates under the same condition. The activity of nuclear factor-kappa B (NFκB) and the expression of its target gene IL-6 were suppressed in SOCS3-knockdown A549 cells and the TG mice after infection with IAV. Moreover, we defined that enhanced STAT3 activity caused by SOCS3 silencing was important for the regulation of NFκB and IL-6. These findings establish a critical role for IL-6-STAT3-SOCS3 axis in the pathogenesis of IAV and suggest that influenza virus may have evolved a strategy to circumvent IL-6/STAT3-mediated immune response through upregulating SOCS3.
甲型流感病毒(IAV)仍然是世界上的主要公共卫生威胁,每年由其感染引起的严重肺炎就是证明。白细胞介素 6(IL-6)参与对 IAV 感染的过度炎症反应,对病毒发病机制有深远影响。然而,这种反应的确切机制尚不清楚。在这里,我们通过 和 研究发现,IAV 不仅诱导了大量的 IL-6 释放,还极大地上调了细胞因子信号转导抑制因子 3(SOCS3)的表达,SOCS3 是 IL-6 相关信号转导和转录激活因子 3(STAT3)信号的有力抑制剂。有趣的是,IAV 在感染的早期阶段存在一种非细胞因子依赖的强大诱导 SOCS3 的机制。此外,我们利用 SOCS3 敲低转基因小鼠(TG)进行了病毒攻击实验,令人惊讶的是,从这些小鼠的实验中观察到,破坏 SOCS3 的表达为 IAV 感染提供了显著的保护,这表现在急性肺损伤减轻、感染动物的存活率提高以及感染组织中的病毒载量降低,与相同条件下的野生型同窝小鼠相比。感染 IAV 后,SOCS3 敲低的 A549 细胞和 TG 小鼠中的核因子-κB(NFκB)活性和其靶基因 IL-6 的表达受到抑制。此外,我们确定了 SOCS3 沉默引起的 STAT3 活性增强对于 NFκB 和 IL-6 的调节很重要。这些发现确立了 IL-6-STAT3-SOCS3 轴在 IAV 发病机制中的关键作用,并表明流感病毒可能已经进化出一种策略,通过上调 SOCS3 来规避 IL-6/STAT3 介导的免疫反应。