Stavropol Research Anti-Plague Institute, 13-15 Sovetskaya Str, Stavropol, Russian Federation, , 355035.
Irkutsk Antiplague Research Institute of Siberia and Far East, Irkutsk, Russian Federation, , 664047.
BMC Genomics. 2019 Sep 2;20(1):692. doi: 10.1186/s12864-019-6060-z.
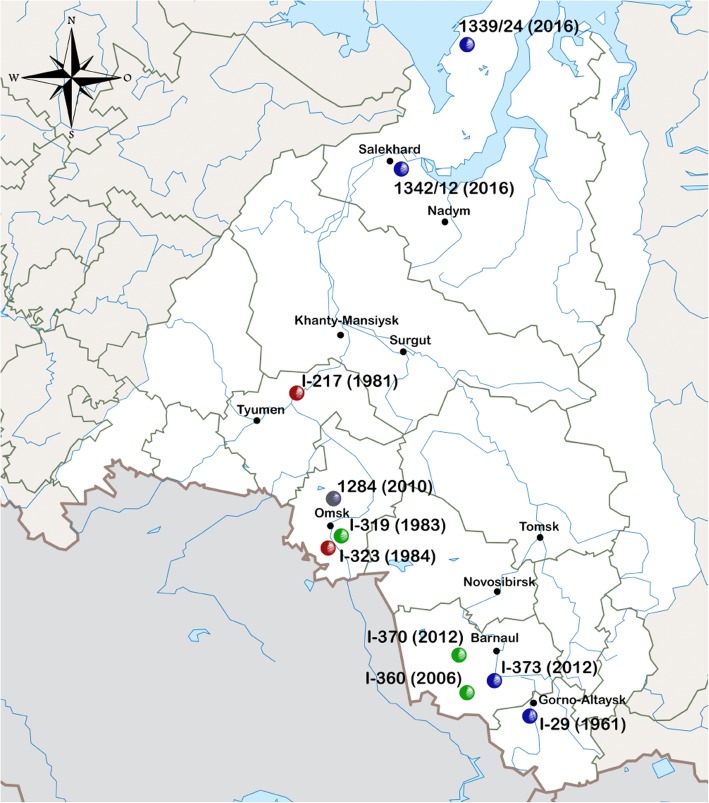
Anthrax is a zoonotic disease caused by the gram-positive bacterium Bacillus anthracis. The most anthrax-endemic regions of Russia are Siberia and North Caucasus. Previously, genotyping of Russian B.anthracis isolates was carried out using canSNP and MLVA data; these methods yield lower resolution results compared to whole genome SNP analysis (wgSNP). In this research, we have used wgSNP method for genotyping of 10 B.anthracis isolates, obtained during 1961-2016 in Russia on territory of Western Siberia.
We have analyzed 185 B.anthracis genomes available in GenBank database and genomes of 10 isolates obtained in this study to determine the place of Russian isolates in the global phylogeny of B.anthracis. For the studied genomes we have detected 7203 SNPs, which were used for building a phylogenetic reconstruction with Maximum Likelihood Method. Results of the phylogenetic analysis indicate that Russian strains belong to three different genetic groups. Three strains belong to genetic group "Ames", two strains - to "STI" group. Five strains belong to the main genetic line B, and four of them form a subcluster, described for the first time, which we have named "Siberia".
In this study, the data on genetic diversity of B.anthracis strains on the territory of Western Siberia is presented for the first time. As a result of complex phylogenetic analysis, the place of these isolates was determined in the global phylogenetic structure of the B.anthracis population. We describe a new cluster in the main genetic line B for the first time.
炭疽是一种由革兰氏阳性细菌炭疽芽孢杆菌引起的人畜共患病。俄罗斯炭疽病流行最严重的地区是西伯利亚和北高加索地区。此前,俄罗斯炭疽芽孢杆菌分离株的基因分型采用了 canSNP 和 MLVA 数据;与全基因组 SNP 分析(wgSNP)相比,这些方法的分辨率较低。在这项研究中,我们使用 wgSNP 方法对 1961 年至 2016 年间在俄罗斯西伯利亚地区获得的 10 株炭疽芽孢杆菌分离株进行了基因分型。
我们分析了 GenBank 数据库中可用的 185 株炭疽芽孢杆菌基因组和本研究中获得的 10 株分离株的基因组,以确定俄罗斯分离株在炭疽芽孢杆菌全球系统发育中的位置。对于研究的基因组,我们检测到了 7203 个 SNP,这些 SNP 用于构建基于最大似然法的系统发育重建。系统发育分析结果表明,俄罗斯菌株属于三个不同的遗传群。三株属于遗传群“Ames”,两株属于“STI”群。五株属于主要遗传谱系 B,其中四株形成了一个亚群,这是首次描述的,我们将其命名为“西伯利亚”。
本研究首次提供了西伯利亚地区炭疽芽孢杆菌菌株遗传多样性的数据。通过复杂的系统发育分析,确定了这些分离株在炭疽芽孢杆菌种群全球系统发育结构中的位置。我们首次描述了主要遗传谱系 B 中的一个新聚类。