Departamento de Bioquímica, Instituto de Química, Universidade de São Paulo, São Paulo, SP, Brazil.
Laboratório Especial de Ciclo Celular, Instituto Butantan, São Paulo, SP, Brazil.
BMC Genomics. 2019 Sep 9;20(1):700. doi: 10.1186/s12864-019-6007-4.
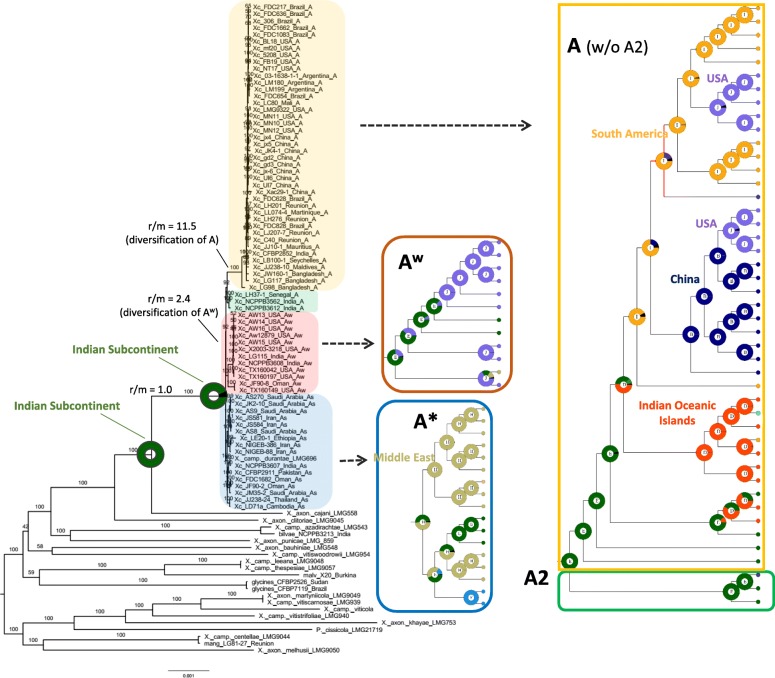
Xanthomonas citri subsp. citri pathotypes cause bacterial citrus canker, being responsible for severe agricultural losses worldwide. The A pathotype has a broad host spectrum, while A* and A are more restricted both in hosts and in geography. Two previous phylogenomic studies led to contrasting well-supported clades for sequenced genomes of these pathotypes. No extensive biogeographical or divergence dating analytic approaches have been so far applied to available genomes.
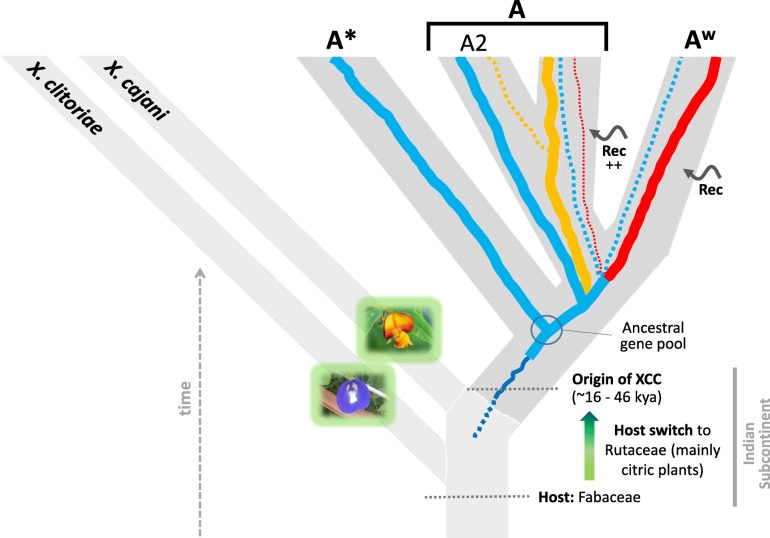
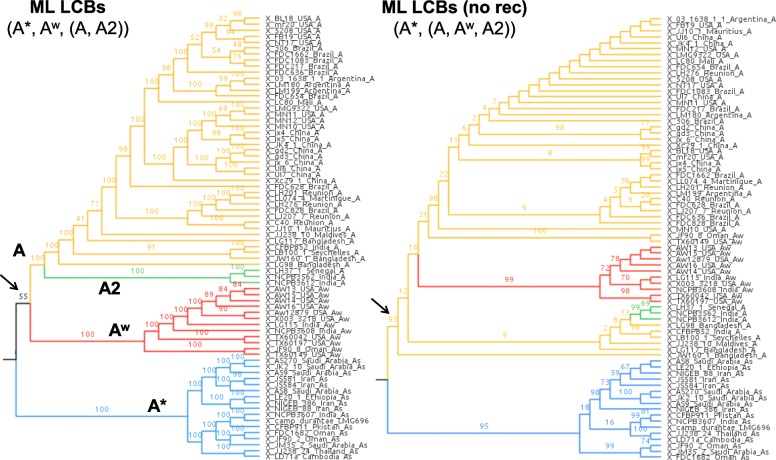
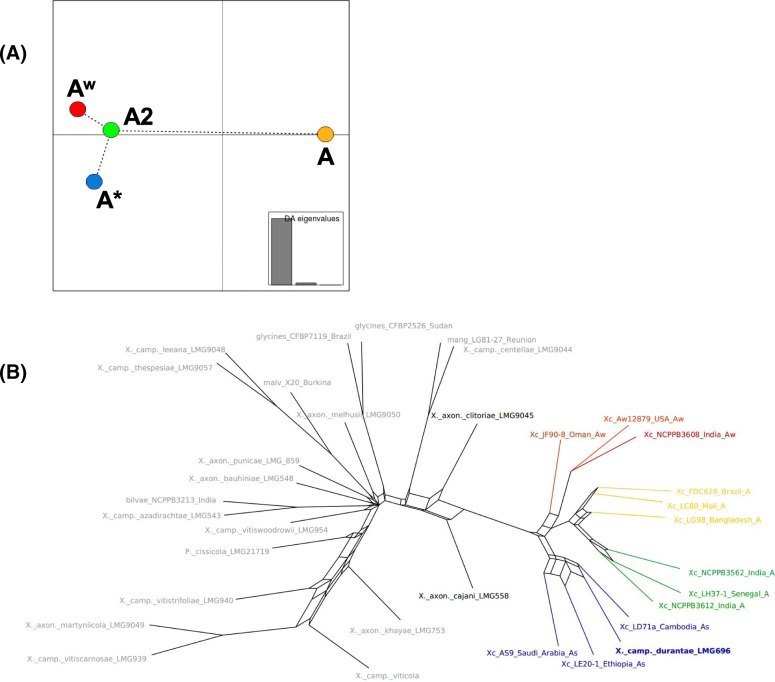
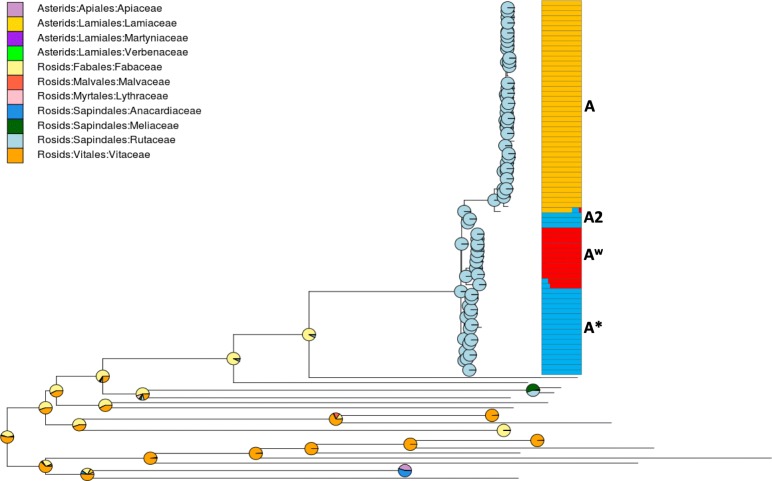
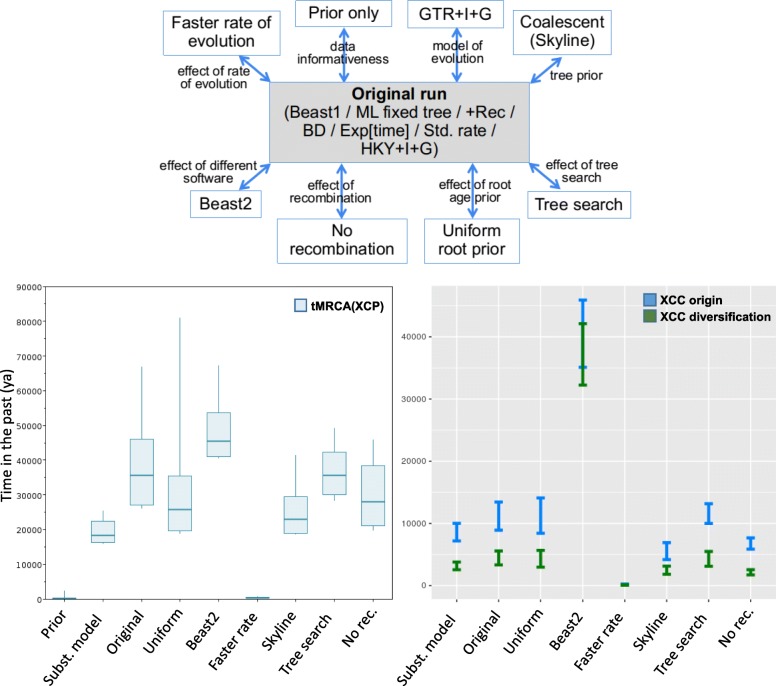
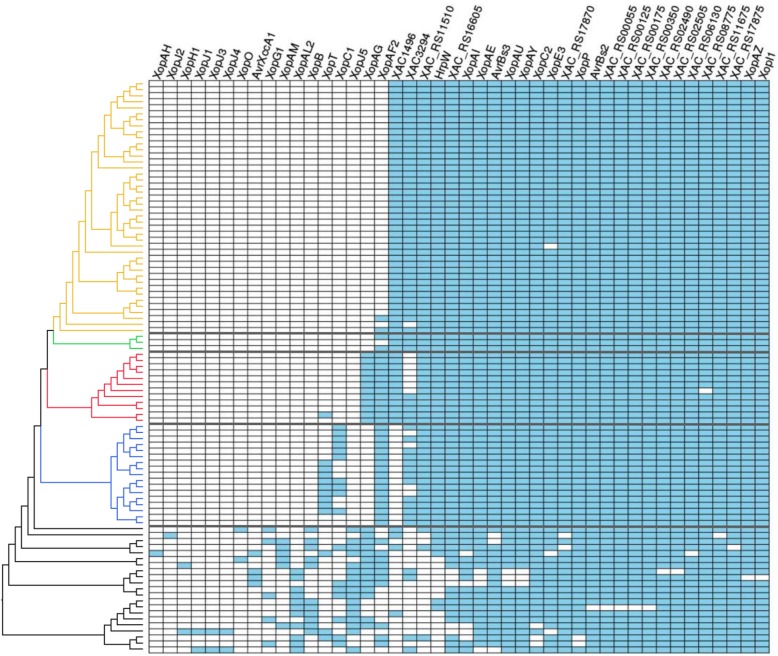
Based on a larger sampling of genomes than in previous studies (including six new genomes sequenced by our group, adding to a total of 95 genomes), phylogenomic analyses resulted in different resolutions, though overall indicating that A + A is the most likely true clade. Our results suggest the high degree of recombination at some branches and the fast diversification of lineages are probable causes for this phylogenetic blurring effect. One of the genomes analyzed, X. campestris pv. durantae, was shown to be an A* strain; this strain has been reported to infect a plant of the family Verbenaceae, though there are no reports of any X. citri subsp. citri pathotypes infecting any plant outside the Citrus genus. Host reconstruction indicated the pathotype ancestor likely had plant hosts in the family Fabaceae, implying an ancient jump to the current Rutaceae hosts. Extensive dating analyses indicated that the origin of X. citri subsp. citri occurred more recently than the main phylogenetic splits of Citrus plants, suggesting dispersion rather than host-directed vicariance as the main driver of geographic expansion. An analysis of 120 pathogenic-related genes revealed pathotype-associated patterns of presence/absence.
Our results provide novel insights into the evolutionary history of X. citri subsp. citri as well as a sound phylogenetic foundation for future evolutionary and genomic studies of its pathotypes.
柑橘黄单胞菌亚种柑橘致病变种引起细菌性柑橘溃疡病,是造成全世界严重农业损失的原因。A 致病变种宿主范围广泛,而 A*和 A 在宿主和地理范围上的限制更为严格。之前的两项基因组系统发育研究导致了对这些致病变种测序基因组的对比支持良好的分支。迄今为止,还没有广泛的生物地理或分歧时间分析方法应用于现有基因组。
基于比以前研究更大的基因组样本(包括我们小组测序的六个新基因组,总共增加到 95 个基因组),基因组系统发育分析得到了不同的分辨率,尽管总体上表明 A+A 是最有可能的真实分支。我们的结果表明,一些分支的高度重组和谱系的快速多样化可能是这种系统发育模糊效应的原因。分析的一个基因组,即野油菜黄单胞菌 pv. 野油菜黄单胞菌,被证明是 A*菌株;据报道,该菌株感染马鞭草科植物,但没有报道任何柑橘黄单胞菌亚种柑橘致病变种感染芸香科以外的任何植物。宿主重建表明,该致病变种的祖先可能有豆科植物作为宿主,这意味着它有一个古老的跳跃到现在的芸香科宿主。广泛的时间分析表明,柑橘黄单胞菌亚种柑橘的起源比柑橘属植物的主要系统发育分支发生得更近,这表明扩散而不是以宿主为导向的地理隔离是其地理扩张的主要驱动因素。对 120 个致病相关基因的分析揭示了存在/缺失的致病变种相关模式。
我们的研究结果为柑橘黄单胞菌亚种柑橘的进化历史提供了新的见解,并为其致病变种的未来进化和基因组研究提供了坚实的系统发育基础。