Gordon Jonathan L, Lefeuvre Pierre, Escalon Aline, Barbe Valérie, Cruveiller Stéphane, Gagnevin Lionel, Pruvost Olivier
Université de la Réunion, UMR PVBMT, 97410, Saint-Pierre, La Réunion, France.
Current Address: CIRAD, UMR CMAEE, F-97170, Petit-Bourg, Guadeloupe, France.
BMC Genomics. 2015 Dec 23;16:1098. doi: 10.1186/s12864-015-2310-x.
The identification of factors involved in the host range definition and evolution is a pivotal challenge in the goal to predict and prevent the emergence of plant bacterial disease. To trace the evolution and find molecular differences between three pathotypes of Xanthomonas citri pv. citri that may explain their distinctive host ranges, 42 strains of X. citri pv. citri and one outgroup strain, Xanthomonas citri pv. bilvae were sequenced and compared.
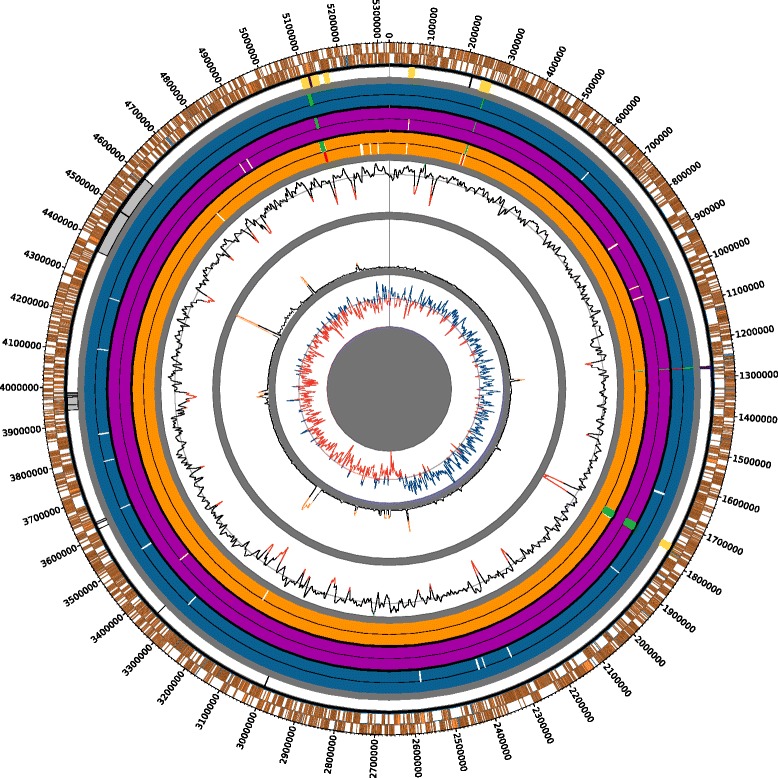
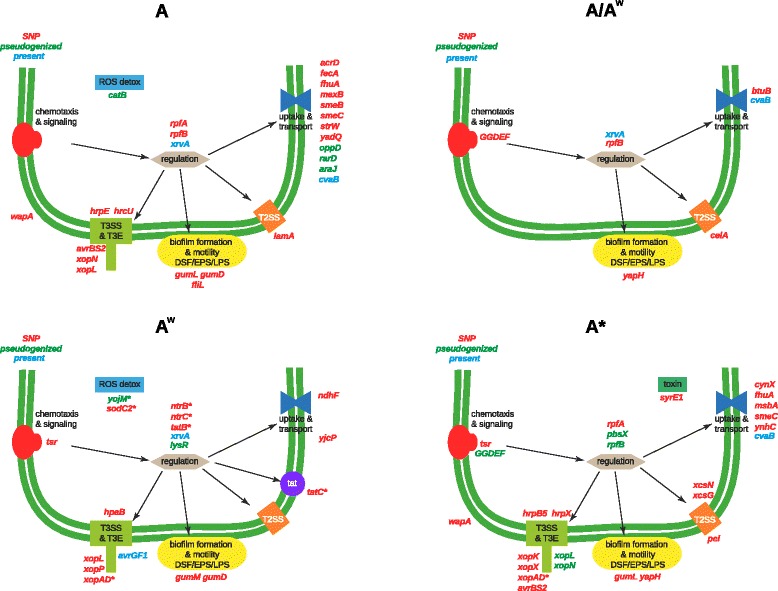
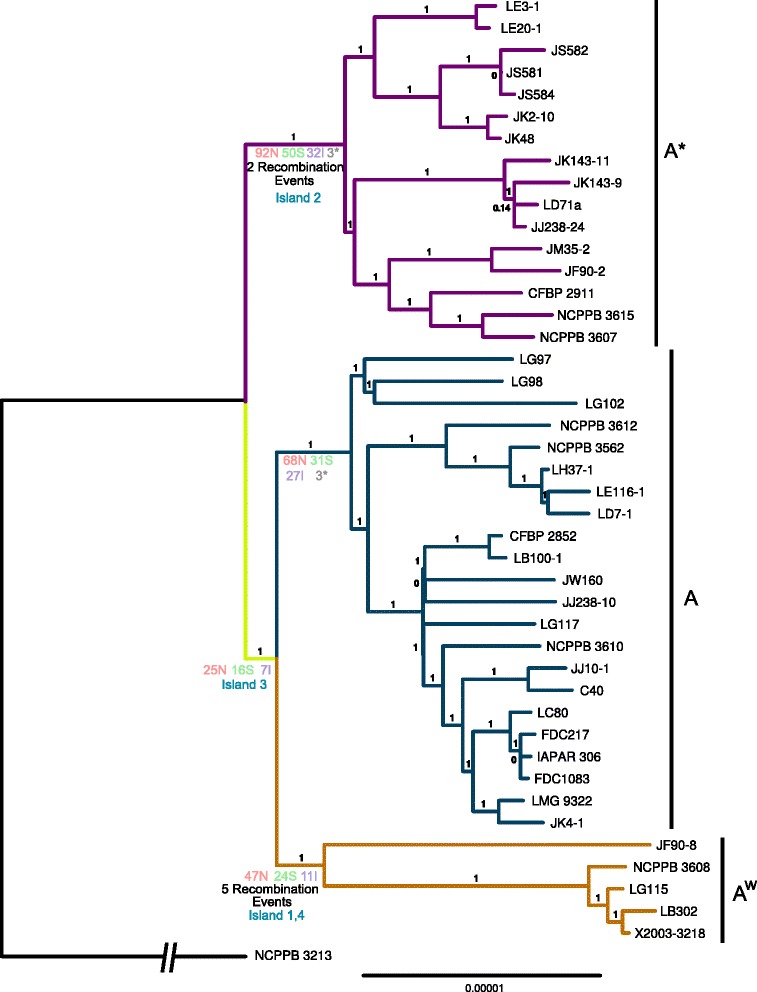
The strains from each pathotype form monophyletic clades, with a short branch shared by the A(w) and A pathotypes. Pathotype-specific recombination was detected in seven regions of the alignment. Using Ancestral Character Estimation, 426 SNPs were mapped to the four branches at the base of the A, A*, A(w) and A/A(w) clades. Several genes containing pathotype-specific nonsynonymous mutations have functions related to pathogenicity. The A pathotype is enriched for SNP-containing genes involved in defense mechanisms, while A* is significantly depleted for genes that are involved in transcription. The pathotypes differ by four gene islands that largely coincide with regions of recombination and include genes with a role in virulence. Both A* and A(w) are missing genes involved in defense mechanisms. In contrast to a recent study, we find that there are an extremely small number of pathotype-specific gene presences and absences.
The three pathotypes of X. citri pv. citri that differ in their host ranges largely show genomic differences related to recombination, horizontal gene transfer and single nucleotide polymorphism. We detail the phylogenetic relationship of the pathotypes and provide a set of candidate genes involved in pathotype-specific evolutionary events that could explain to the differences in host range and pathogenicity between them.
确定参与宿主范围界定和进化的因素是预测和预防植物细菌病害出现这一目标中的关键挑战。为了追踪进化过程并找出柑橘溃疡病菌三个致病型之间可能解释其独特宿主范围的分子差异,对42株柑橘溃疡病菌和一株外类群菌株——野油菜黄单胞菌进行了测序和比较。
每个致病型的菌株形成单系分支,A(w)和A致病型共有一个短分支。在比对的七个区域检测到致病型特异性重组。使用祖先特征估计,426个单核苷酸多态性(SNP)被定位到A、A*、A(w)和A/A(w)分支基部的四个分支上。几个含有致病型特异性非同义突变的基因具有与致病性相关的功能。A致病型富含参与防御机制的含SNP基因,而A中参与转录的基因显著减少。这些致病型在四个基因岛方面存在差异,这些基因岛在很大程度上与重组区域重合,并且包括在毒力方面起作用的基因。A和A(w)都缺失参与防御机制的基因。与最近的一项研究相反,我们发现致病型特异性基因的存在和缺失数量极少。
宿主范围不同的柑橘溃疡病菌的三个致病型在很大程度上表现出与重组、水平基因转移和单核苷酸多态性相关的基因组差异。我们详细阐述了致病型的系统发育关系,并提供了一组参与致病型特异性进化事件的候选基因,这些事件可以解释它们之间宿主范围和致病性的差异。