Seth-Smith Helena M B, Bonfiglio Ferdinando, Cuénod Aline, Reist Josiane, Egli Adrian, Wüthrich Daniel
Division of Clinical Bacteriology and Mycology, University Hospital Basel, Basel, Switzerland.
Applied Microbiology Research, Department of Biomedicine, University of Basel, Basel, Switzerland.
Front Public Health. 2019 Aug 27;7:241. doi: 10.3389/fpubh.2019.00241. eCollection 2019.
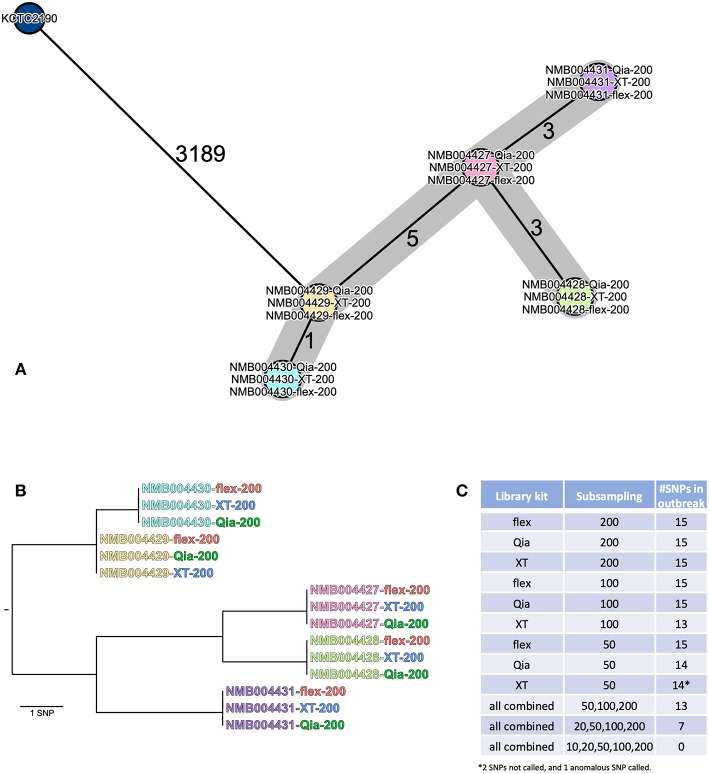
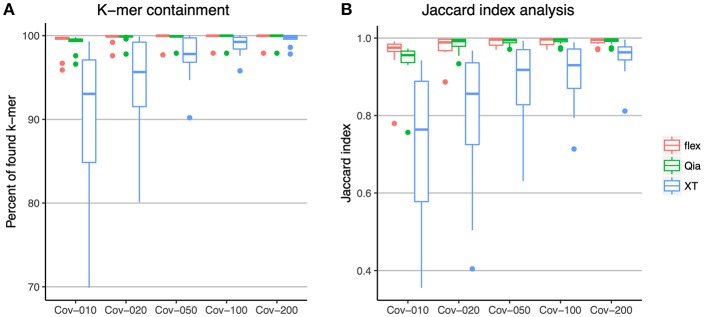
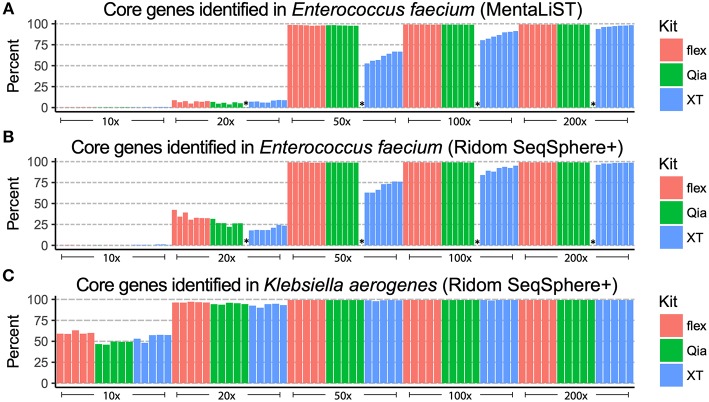
Whole genome sequencing (WGS) has become the new gold standard for bacterial outbreak investigation, due to the high resolution available for typing. While sequencing is currently predominantly performed on Illumina devices, the preceding library preparation can be performed using various protocols. Enzymatic fragmentation library preparation protocols are fast, have minimal hands-on time, and work with small quantities of DNA. The aim of our study was to compare three library preparation protocols for molecular typing: Nextera XT (Illumina); Nextera Flex (Illumina); and QIAseq FX (Qiagen). We selected 12 ATCC strains from human Gram-positive and Gram-negative pathogens with %G+C-content ranging from 27% () to 73% (), each having a high quality complete genome assembly available, to allow in-depth analysis of the resulting Illumina sequence data quality. Additionally, we selected isolates from previously analyzed cases of vancomycin-resistant (VRE) ( = 7) and a local outbreak of ( = 5). The number of protocol steps and time required were compared, in order to test the suitability for routine laboratory work. Data analyses were performed with standard tools commonly used in outbreak situations: Ridom SeqSphere+ for cgMLST; CLC genomics workbench for SNP analysis; and open source programs. Nextera Flex and QIAseq FX were found to be less sensitive than Nextera XT to variable %G+C-content, resulting in an almost uniform distribution of read-depth. Therefore, low coverage regions are reduced to a minimum resulting in a more complete representation of the genome. Thus, with these two protocols, more alleles were detected in the cgMLST analysis, producing a higher resolution of closely related isolates. Furthermore, they result in a more complete representation of accessory genes. In particular, the high data quality and relative simplicity of the workflow of Nextera Flex stood out in this comparison. This thorough comparison within an ISO/IEC 17025 accredited environment will be of interest to those aiming to optimize their clinical microbiological genome sequencing.
由于全基因组测序(WGS)在分型方面具有高分辨率,已成为细菌暴发调查的新金标准。虽然目前测序主要在Illumina设备上进行,但先前的文库制备可以使用各种方案。酶切片段化文库制备方案速度快、实际操作时间最少,并且适用于少量DNA。我们研究的目的是比较三种用于分子分型的文库制备方案:Nextera XT(Illumina);Nextera Flex(Illumina);以及QIAseq FX(Qiagen)。我们从人类革兰氏阳性和革兰氏阴性病原体中选择了12株ATCC菌株,其G+C含量范围为27%( )至73%( ),每株菌株都有高质量的完整基因组组装,以便深入分析所得Illumina序列数据的质量。此外,我们从先前分析的耐万古霉素肠球菌(VRE)病例(n = 7)和当地的一次肠球菌暴发(n = 5)中选择了分离株。比较了方案步骤的数量和所需时间,以测试其是否适合常规实验室工作。使用暴发情况下常用的标准工具进行数据分析:用于cgMLST的Ridom SeqSphere+;用于SNP分析的CLC基因组学工作台;以及开源程序。发现Nextera Flex和QIAseq FX比Nextera XT对可变的G+C含量不太敏感,导致读深度几乎均匀分布。因此,低覆盖区域减少到最低限度,从而更完整地呈现基因组。因此,使用这两种方案,在cgMLST分析中检测到更多等位基因,对密切相关的分离株产生更高的分辨率。此外,它们能更完整地呈现辅助基因。特别是,Nextera Flex工作流程的数据质量高且相对简单,在这次比较中脱颖而出。对于那些旨在优化其临床微生物基因组测序的人来说,在ISO/IEC 17025认可环境内进行的这种全面比较将很有意义。