Li Guilan, Gao Guanjie, Wang Panfeng, Song Xiaojing, Xu Ping, Xie Bingbing, Zhou Tiancheng, Pan Guangjin, Peng Fuhua, Zhang Qingjiong, Ge Jian, Zhong Xiufeng
State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China.
Key Laboratory of Regenerative Biology, South China Institute for Stem Cell Biology and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China.
Front Mol Neurosci. 2019 Sep 11;12:212. doi: 10.3389/fnmol.2019.00212. eCollection 2019.
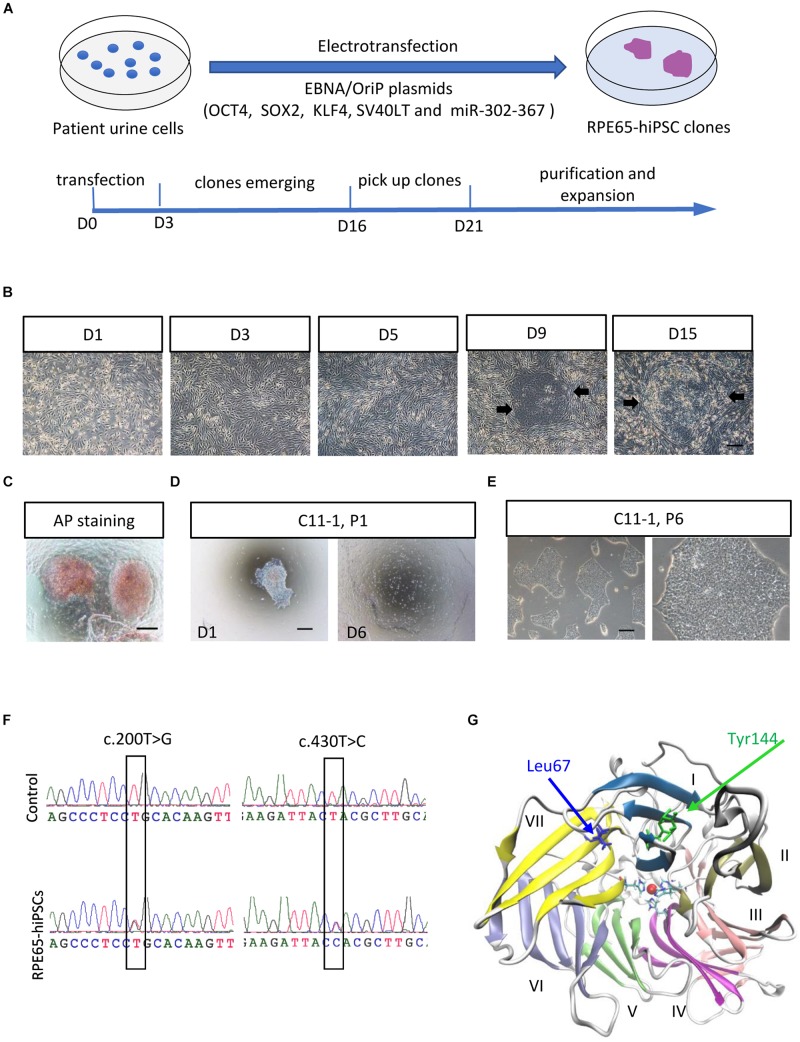
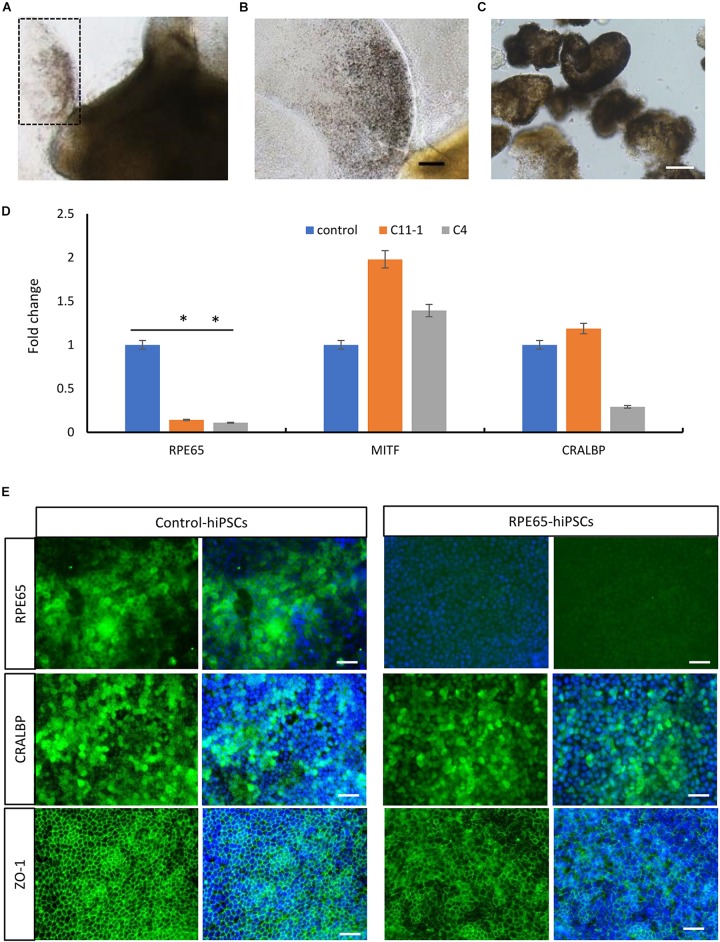
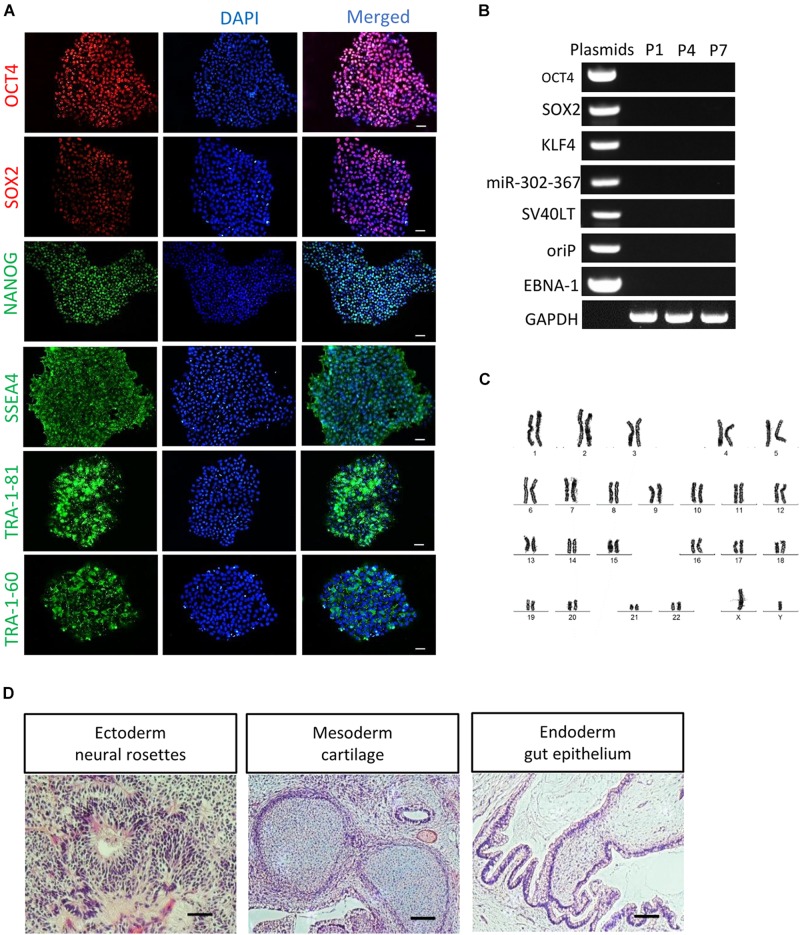
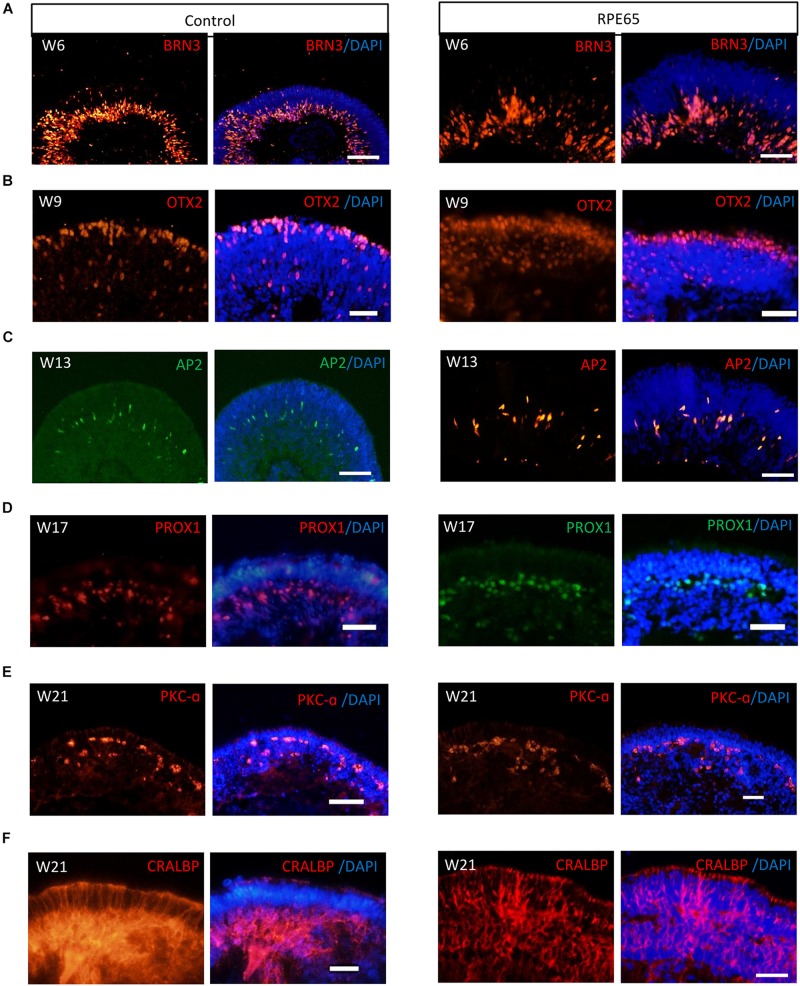
RPE65-associated Leber congenital amaurosis (LCA) is one of highly heterogeneous, early onset, severe retinal dystrophies with at least 130 gene mutation sites identified. Their pathogenicity has not been directly clarified due to lack of diseased cells. Here, we generated human-induced pluripotent stem cells (hiPSCs) from one putative LCA patient carrying two novel mutations with c.200T>G (p.L67R) and c.430T>C (p.Y144H), named RPE65-hiPSCs, which were confirmed to contain the same mutations. The RPE65-hiPSCs presented typical morphological features with normal karyotype, expressed pluripotency markers, and developed teratoma in NOD-SCID mice. Moreover, the patient hiPSCs were able to differentiate toward retinal lineage fate and self-form retinal organoids with layered neural retina. All major retinal cell types including photoreceptor and retinal pigment epithelium (RPE) cells were also acquired overtime. Compared to healthy control, RPE cells from patient iPSCs had lower expression of RPE65, but similar phagocytic activity and VEGF secretion level. This study provided the valuable patient specific, disease targeted retinal organoids containing photoreceptor and RPE cells, which would facilitate the study of personalized pathogenic mechanisms of disease, drug screening, and cell replacement therapy.
视网膜色素上皮特异性 65 蛋白相关的莱伯先天性黑蒙(LCA)是一种高度异质性、早发性、严重的视网膜营养不良,已确定至少有 130 个基因突变位点。由于缺乏病变细胞,它们的致病性尚未得到直接阐明。在此,我们从一名疑似 LCA 患者中生成了携带两个新突变 c.200T>G(p.L67R)和 c.430T>C(p.Y144H)的人诱导多能干细胞(hiPSC),命名为 RPE65-hiPSC,经证实其含有相同的突变。RPE65-hiPSC 呈现出典型的形态特征,核型正常,表达多能性标志物,并在 NOD-SCID 小鼠中形成畸胎瘤。此外,患者的 hiPSC 能够向视网膜谱系命运分化,并自我形成具有分层神经视网膜的视网膜类器官。随着时间的推移,还获得了包括光感受器和视网膜色素上皮(RPE)细胞在内的所有主要视网膜细胞类型。与健康对照相比,患者 iPSC 来源的 RPE 细胞中 RPE65 的表达较低,但吞噬活性和 VEGF 分泌水平相似。本研究提供了有价值的患者特异性、疾病靶向性的包含光感受器和 RPE 细胞的视网膜类器官,这将有助于研究疾病的个性化致病机制、药物筛选和细胞替代治疗。