Neurometabolic Diseases Laboratory, Bellvitge Biomedical Research Institute (IDIBELL), L'Hospitalet de Llobregat, Barcelona, Catalunya, Spain.
Centre for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Barcelona, Spain.
J Med Genet. 2020 Feb;57(2):132-137. doi: 10.1136/jmedgenet-2019-106373. Epub 2019 Oct 5.
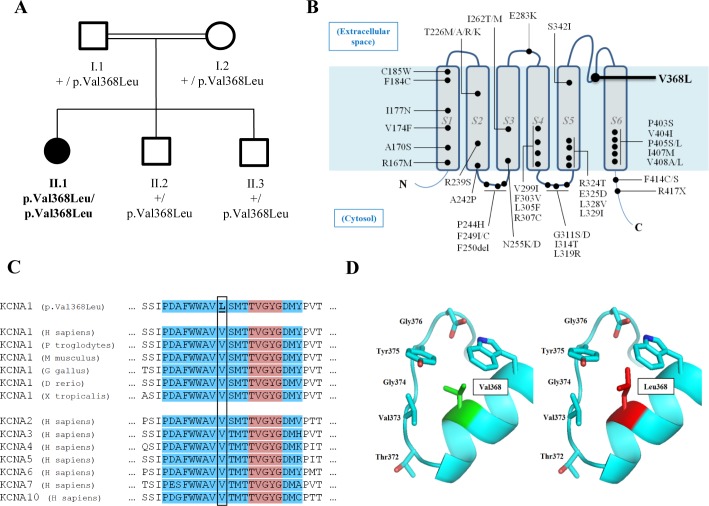
Since 1994, over 50 families affected by the episodic ataxia type 1 disease spectrum have been described with mutations in , encoding the voltage-gated K channel subunit Kv1.1. All of these mutations are either transmitted in an autosomal-dominant mode or found as events.
A patient presenting with a severe combination of dyskinesia and neonatal epileptic encephalopathy was sequenced by whole-exome sequencing (WES). A candidate variant was tested using cellular assays and patch-clamp recordings.
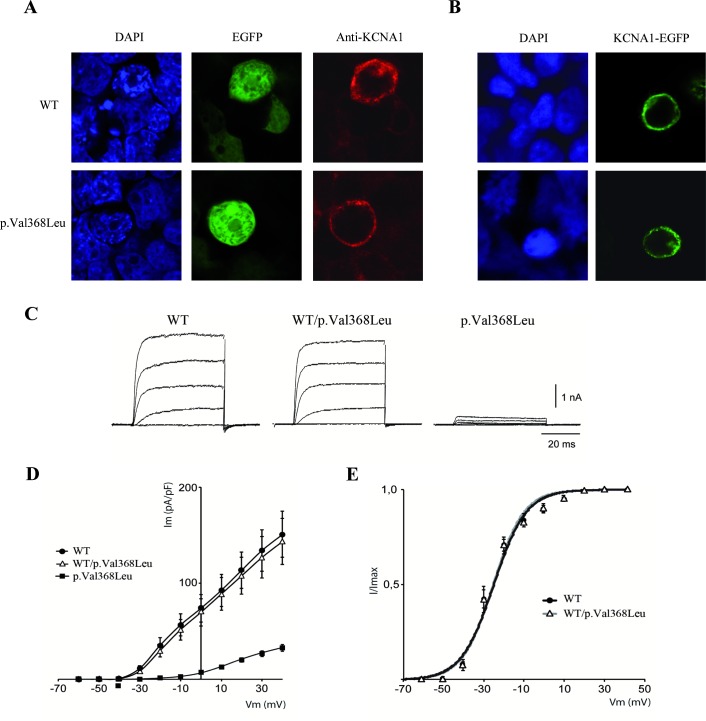
WES revealed a homozygous variant (p.Val368Leu) in , involving a conserved residue in the pore domain, close to the selectivity signature sequence for K ions (TVGYG). Functional analysis showed that mutant protein alone failed to produce functional channels in homozygous state, while coexpression with wild-type produced no effects on K currents, similar to wild-type protein alone. Treatment with oxcarbazepine, a sodium channel blocker, proved effective in controlling seizures.
This newly identified variant is the first to be reported to act in a recessive mode of inheritance in . These findings serve as a cautionary tale for the diagnosis of channelopathies, in which an unreported phenotypic presentation or mode of inheritance for the variant of interest can hinder the identification of causative variants and adequate treatment choice.
自 1994 年以来,已有超过 50 个受发作性共济失调 1 型疾病谱影响的家族被描述,这些家族的基因突变都位于 基因上,该基因编码电压门控钾通道亚单位 Kv1.1。所有这些突变要么以常染色体显性模式遗传,要么以纯合缺失的形式出现。
通过全外显子组测序(WES)对一名表现出严重运动障碍和新生儿癫痫脑病的患者进行测序。使用细胞检测和膜片钳记录来测试候选变体。
WES 揭示了 基因中的一个纯合变异(p.Val368Leu),该变异涉及孔域中的保守残基,靠近钾离子选择性特征序列(TVGYG)。功能分析表明,突变蛋白在纯合状态下单独无法产生功能性通道,而与野生型共表达对 K 电流没有影响,与单独的野生型蛋白相似。奥卡西平(一种钠离子通道阻滞剂)的治疗对控制癫痫发作有效。
这个新发现的变异是首次报道在 基因中以隐性遗传模式起作用。这些发现为通道病的诊断敲响了警钟,对于感兴趣的变异,未报告的表型表现或遗传模式可能会阻碍致病变异的识别和适当的治疗选择。