Karalok Zeynep S, Megaro Alfredo, Cenciarini Marta, Guven Alev, Hasan Sonia M, Taskin Birce D, Imbrici Paola, Ceylaner Serdar, Pessia Mauro, D'Adamo Maria C
Department of Pediatric Neurology, Ankara Children's Hematology Oncology Research and Training Hospital, Ankara, Turkey.
Section of Physiology and Biochemistry, Department of Experimental Medicine, School of Medicine, University of Perugia, Perugia, Italy.
Front Neurol. 2018 Jul 25;9:587. doi: 10.3389/fneur.2018.00587. eCollection 2018.
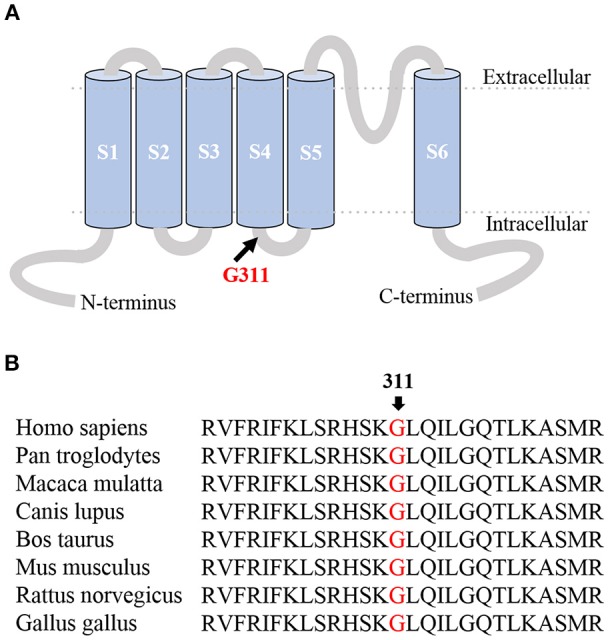
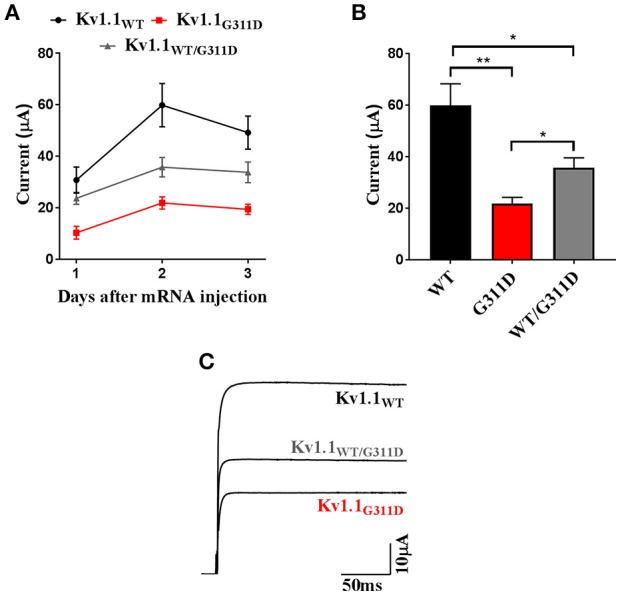
Episodic ataxia type 1 (EA1), a -like K, is a consequence of genetic anomalies in the gene that lead to dysfunctions in the voltage-gated K channel Kv1. 1. Generally, mutations are inherited in an autosomal dominant manner. Here we report the clinical phenotype of an EA1 patient characterized by ataxia attacks that decrease in frequency with age, and eventually leading to therapy discontinuation. A new mutation (c.932G>A) that changed a highly conserved glycine residue into an aspartate (p.G311D) was identified by using targeted next-generation sequencing. The conserved glycine is located in the S4-S5 linker, a crucial domain controlling Kv1.1 channel gating. analyses predicted the mutation deleterious. Heterologous expression of the mutant (Kv1.1-G311D) channels resulted in remarkably decreased amplitudes of measured current, confirming the identified variant is pathogenic. Collectively, these findings corroborate the notion that EA1 also results from variants and point out that regardless of the mutation-induced deleterious loss of Kv1.1 channel function the ataxia phenotype may improve spontaneously.
发作性共济失调1型(EA1),一种类似α样的K,是由基因中的遗传异常导致电压门控钾通道Kv1.1功能障碍的结果。一般来说,突变以常染色体显性方式遗传。在此我们报告一名EA1患者的临床表型,其特征为共济失调发作频率随年龄降低,最终导致治疗中断。通过靶向新一代测序鉴定出一个新的突变(c.932G>A),该突变将一个高度保守的甘氨酸残基变为天冬氨酸(p.G311D)。保守的甘氨酸位于S4 - S5连接区,这是控制Kv1.1通道门控的关键结构域。分析预测该突变有害。突变型(Kv1.1 - G311D)通道的异源表达导致测量电流幅度显著降低,证实所鉴定的变异是致病的。总体而言,这些发现证实了EA1也由变异导致的观点,并指出无论突变引起的Kv1.1通道功能有害丧失如何,共济失调表型可能会自发改善。