The Milner Centre for Evolution, Department of Biology and Biochemistry, Claverton Down, University of Bath, Bath BA2 7AY, UK.
Gigascience. 2019 Oct 1;8(10). doi: 10.1093/gigascience/giz119.
Cataloguing the distribution of genes within natural bacterial populations is essential for understanding evolutionary processes and the genetic basis of adaptation. Advances in whole genome sequencing technologies have led to a vast expansion in the amount of bacterial genomes deposited in public databases. There is a pressing need for software solutions which are able to cluster, catalogue and characterise genes, or other features, in increasingly large genomic datasets.
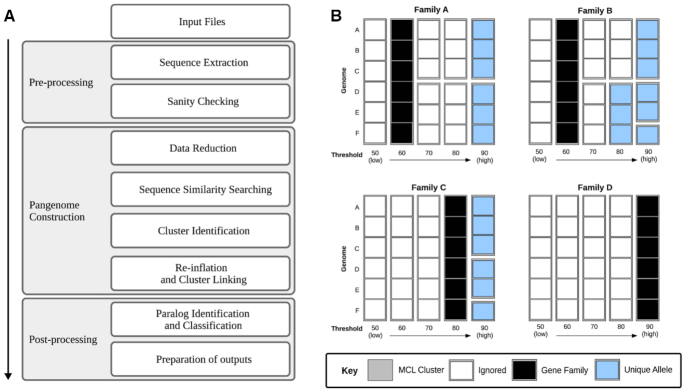
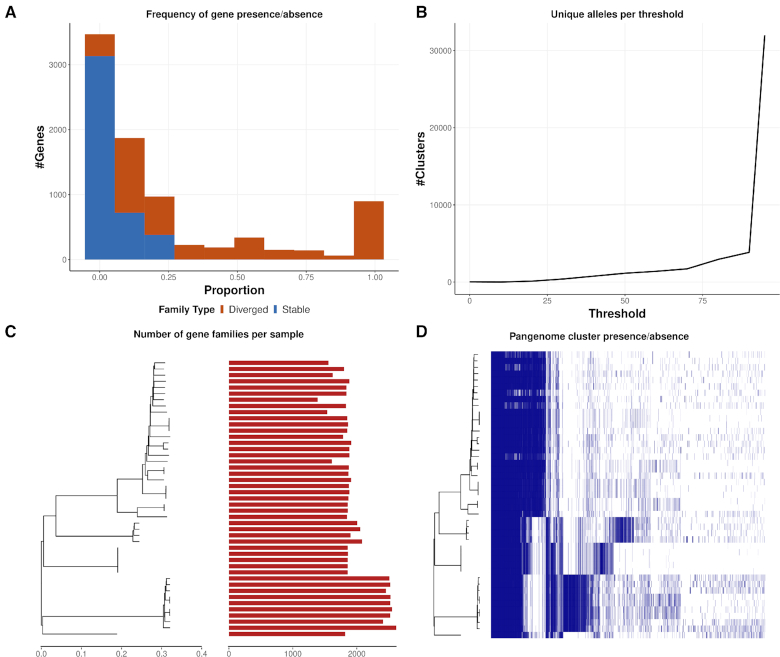
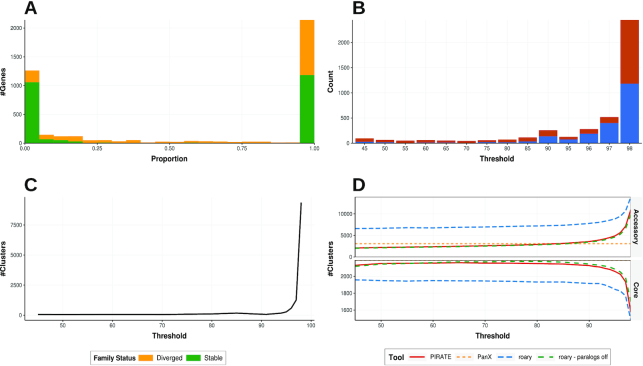
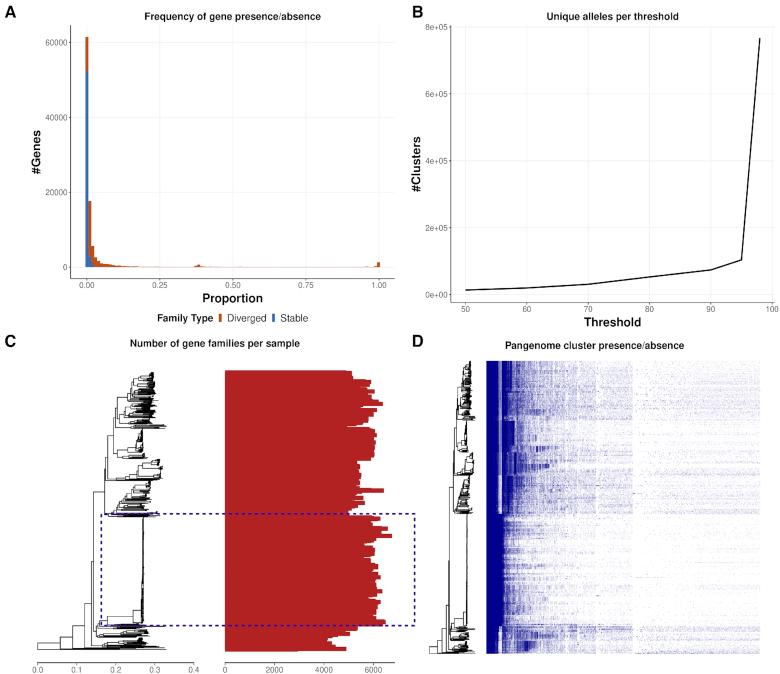
Here we present a pangenomics toolbox, PIRATE (Pangenome Iterative Refinement and Threshold Evaluation), which identifies and classifies orthologous gene families in bacterial pangenomes over a wide range of sequence similarity thresholds. PIRATE builds upon recent scalable software developments to allow for the rapid interrogation of thousands of isolates. PIRATE clusters genes (or other annotated features) over a wide range of amino acid or nucleotide identity thresholds and uses the clustering information to rapidly identify paralogous gene families and putative fission/fusion events. Furthermore, PIRATE orders the pangenome using a directed graph, provides a measure of allelic variation, and estimates sequence divergence for each gene family.
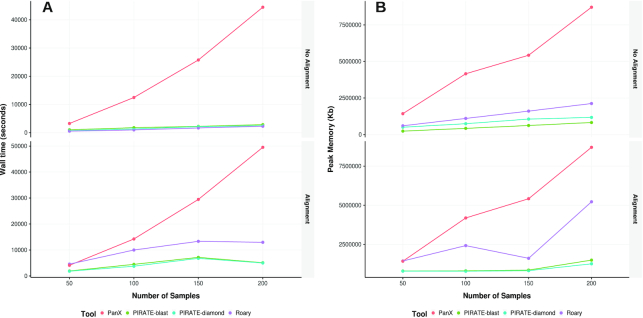
We demonstrate that PIRATE scales linearly with both number of samples and computation resources, allowing for analysis of large genomic datasets, and compares favorably to other popular tools. PIRATE provides a robust framework for analysing bacterial pangenomes, from largely clonal to panmictic species.
编目自然细菌种群中基因的分布对于理解进化过程和适应的遗传基础至关重要。全基因组测序技术的进步导致公共数据库中储存的细菌基因组数量大幅增加。现在迫切需要能够对越来越大的基因组数据集进行聚类、编目和特征分析的软件解决方案。
在这里,我们提出了一个泛基因组工具包 PIRATE(泛基因组迭代细化和阈值评估),它可以在广泛的序列相似性阈值范围内识别和分类细菌泛基因组中的直系同源基因家族。PIRATE 建立在最近的可扩展软件发展的基础上,允许快速询问数千个分离物。PIRATE 在广泛的氨基酸或核苷酸身份阈值范围内对基因(或其他注释特征)进行聚类,并使用聚类信息快速识别旁系基因家族和潜在的分裂/融合事件。此外,PIRATE 使用有向图对泛基因组进行排序,提供等位基因变异的度量,并估计每个基因家族的序列分歧。
我们证明 PIRATE 可以线性扩展到样本数量和计算资源,允许对大型基因组数据集进行分析,并与其他流行工具相比具有优势。PIRATE 为分析从主要克隆到泛化的细菌泛基因组提供了一个稳健的框架。