Lin Guanyu, Yin Guoqian, Yan Yuyong, Lin Bojie
Department of Plastic and Aesthetic Surgery, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, P.R. China.
Oncol Lett. 2019 Nov;18(5):5243-5254. doi: 10.3892/ol.2019.10914. Epub 2019 Sep 24.
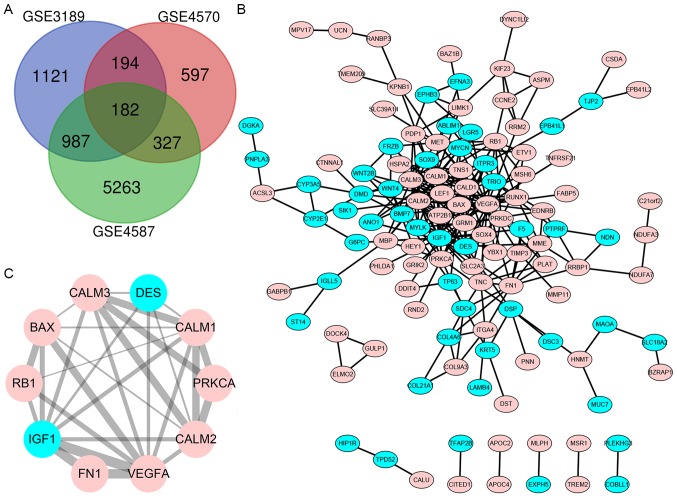
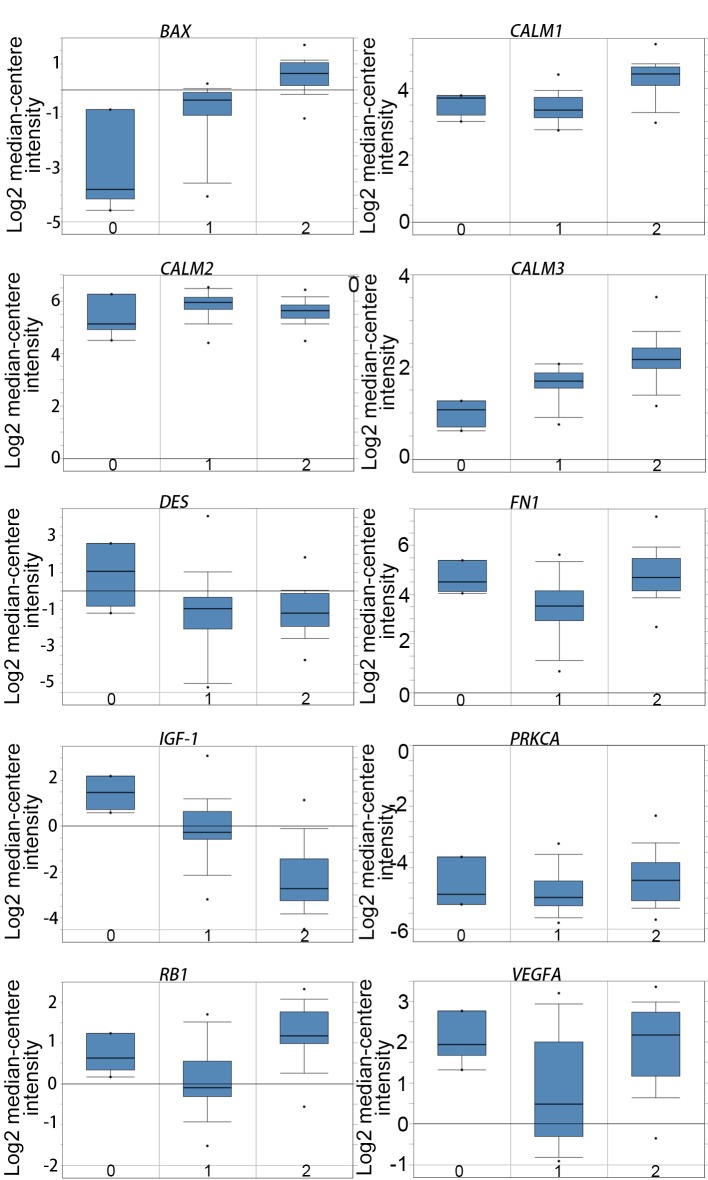
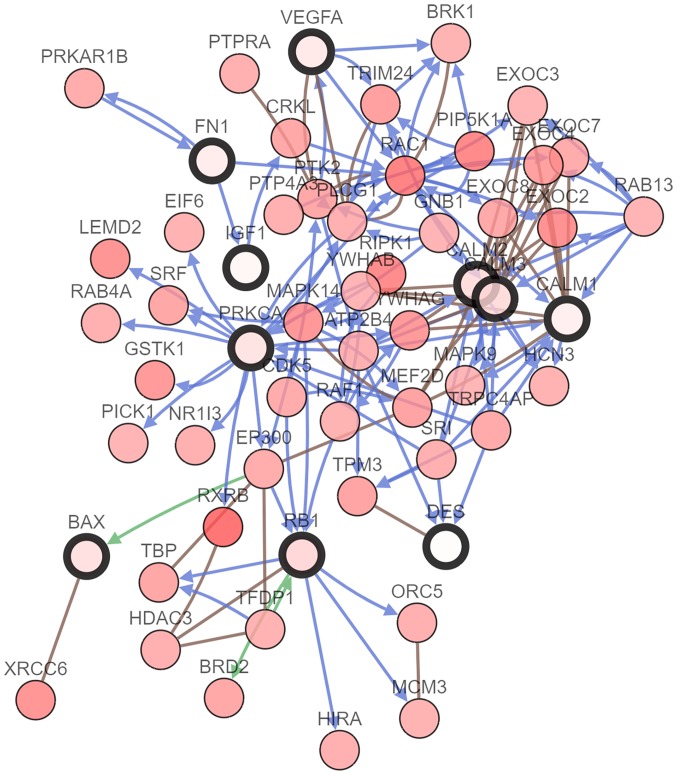
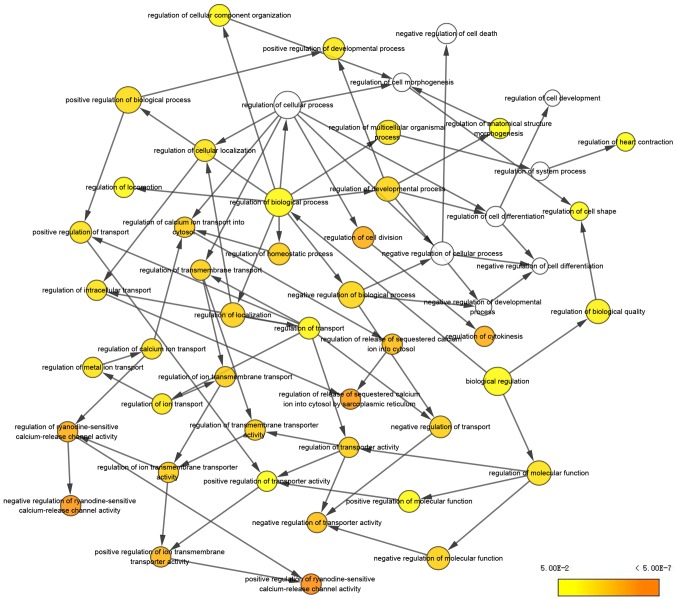
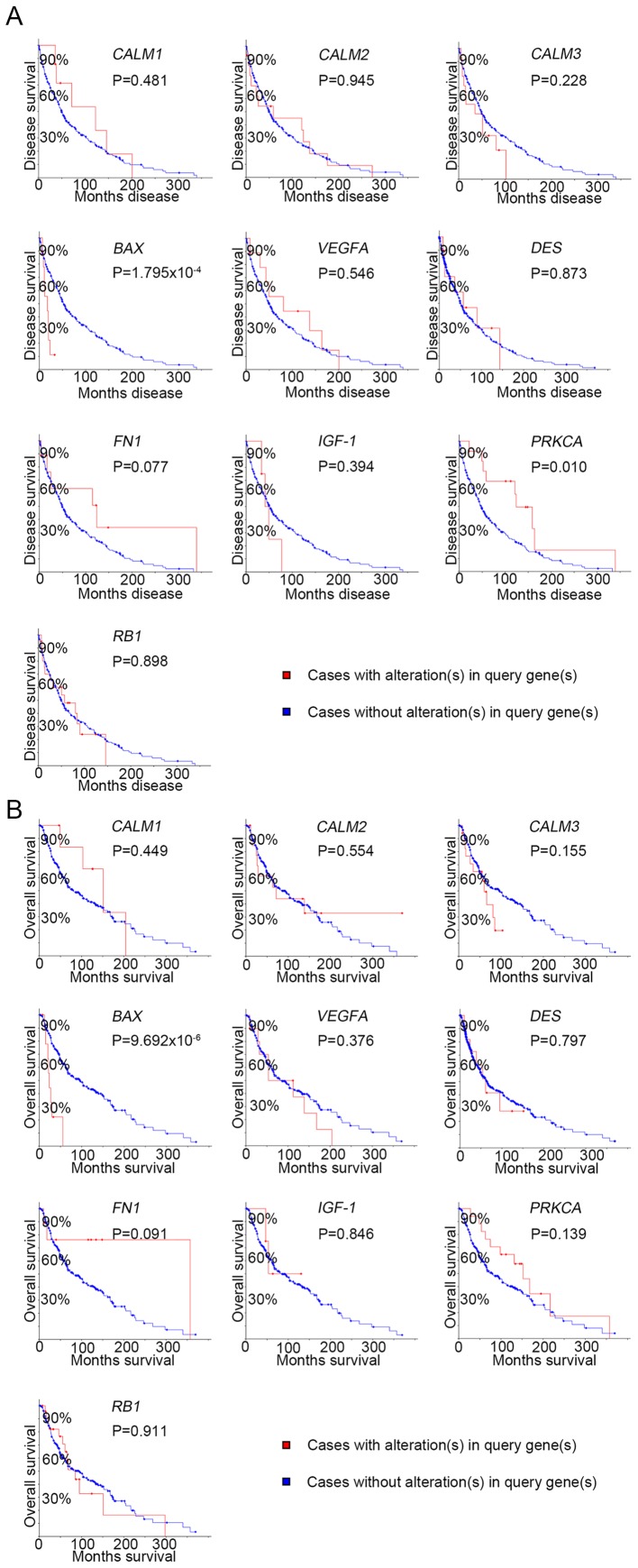
Malignant melanoma is one of the most common types of cancer worldwide. Efforts have been made to elucidate the pathology of malignant melanoma. However, its molecular mechanisms remain unclear. Therefore, the microarray datasets GSE3189, GSE4570 and GSE4587 from the Gene Expression Omnibus database were used for the elucidation of candidate genes involved in the initiation and progression of melanoma. Assessment of the microarray datasets led to the identification of differentially expressed genes (DEGs), which were subsequently used for function enrichment analysis. These data were utilized in the construction of the protein-protein interaction network and module analysis was conducted using STRING and Cytoscape software. The results of these analyses led to the identification of a total of 182 DEGs, including 52 downregulated and 130 upregulated genes. The functions and pathways found to be enriched in the DEGs were GTPase activity, transcription from RNA polymerase II promoter, apoptotic processes, cell adhesion, membrane related pathways, calcium signaling cascade and the PI3K-Akt signaling pathway. The identified genes were demonstrated to belong to a set of 10 hub genes biologically involved in proliferation, apoptosis, cytokinesis, adhesion and migration. Survival analysis and Oncomine database analysis revealed that the calmodulin gene family, and genes, may be associated with the initiation, invasion or recurrence of melanoma. In conclusion, the DEGs and hub genes identified in the present study may be used to understand the molecular pathways involved in the initiation and progression of malignant melanoma. Furthermore, the present study may aid in the identification of possible targets for the diagnosis and treatment of melanoma.
恶性黑色素瘤是全球最常见的癌症类型之一。人们已努力阐明恶性黑色素瘤的病理学。然而,其分子机制仍不清楚。因此,使用来自基因表达综合数据库的微阵列数据集GSE3189、GSE4570和GSE4587来阐明参与黑色素瘤发生和发展的候选基因。对微阵列数据集的评估导致了差异表达基因(DEG)的鉴定,随后将这些基因用于功能富集分析。这些数据被用于构建蛋白质-蛋白质相互作用网络,并使用STRING和Cytoscape软件进行模块分析。这些分析结果共鉴定出182个DEG,包括52个下调基因和130个上调基因。在DEG中发现富集的功能和途径包括GTPase活性、RNA聚合酶II启动子的转录、凋亡过程、细胞粘附、膜相关途径、钙信号级联和PI3K-Akt信号通路。已证明所鉴定的基因属于一组在生物学上参与增殖、凋亡、胞质分裂、粘附和迁移的10个枢纽基因。生存分析和Oncomine数据库分析表明,钙调蛋白基因家族以及其他基因可能与黑色素瘤的发生、侵袭或复发有关。总之,本研究中鉴定的DEG和枢纽基因可用于了解恶性黑色素瘤发生和发展所涉及的分子途径。此外,本研究可能有助于确定黑色素瘤诊断和治疗的潜在靶点。