Jones Katy E, Fér Tomáš, Schmickl Roswitha E, Dikow Rebecca B, Funk Vicki A, Herrando-Moraira Sonia, Johnston Paul R, Kilian Norbert, Siniscalchi Carolina M, Susanna Alfonso, Slovák Marek, Thapa Ramhari, Watson Linda E, Mandel Jennifer R
Botanischer Garten und Botanisches Museum Berlin Freie Universität Berlin Königin-Luise-Str. 6-8 14195 Berlin Germany.
Department of Botany Faculty of Science Charles University Benátská 2 CZ 12800 Prague Czech Republic.
Appl Plant Sci. 2019 Oct 25;7(10):e11295. doi: 10.1002/aps3.11295. eCollection 2019 Oct.
Hybrid capture with high-throughput sequencing (Hyb-Seq) is a powerful tool for evolutionary studies. The applicability of an Asteraceae family-specific Hyb-Seq probe set and the outcomes of different phylogenetic analyses are investigated here.
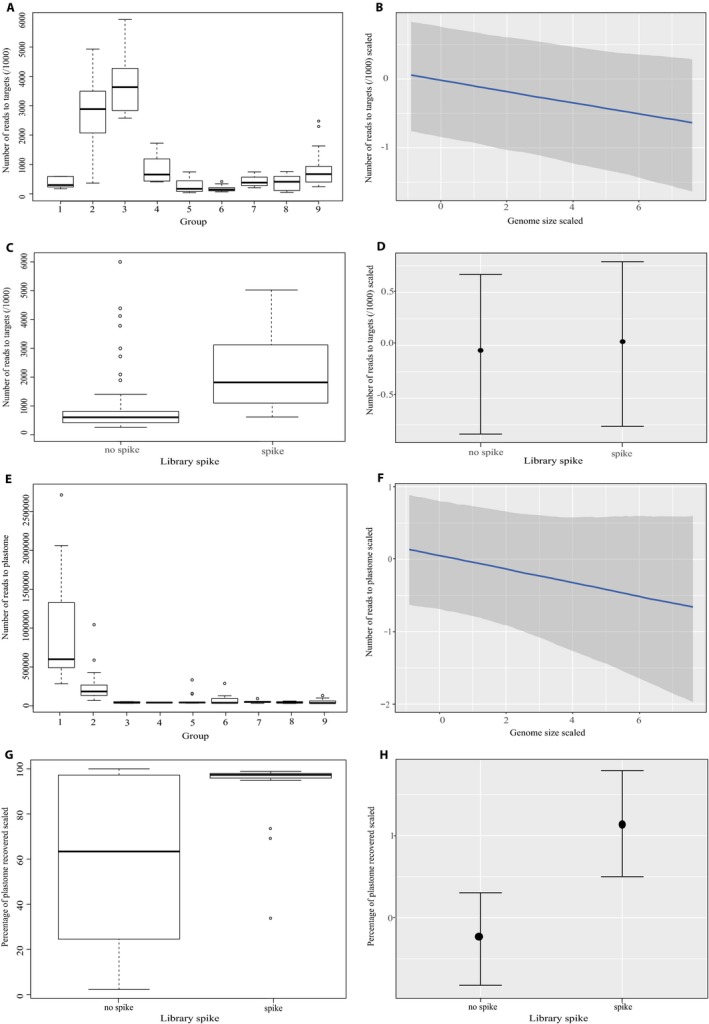
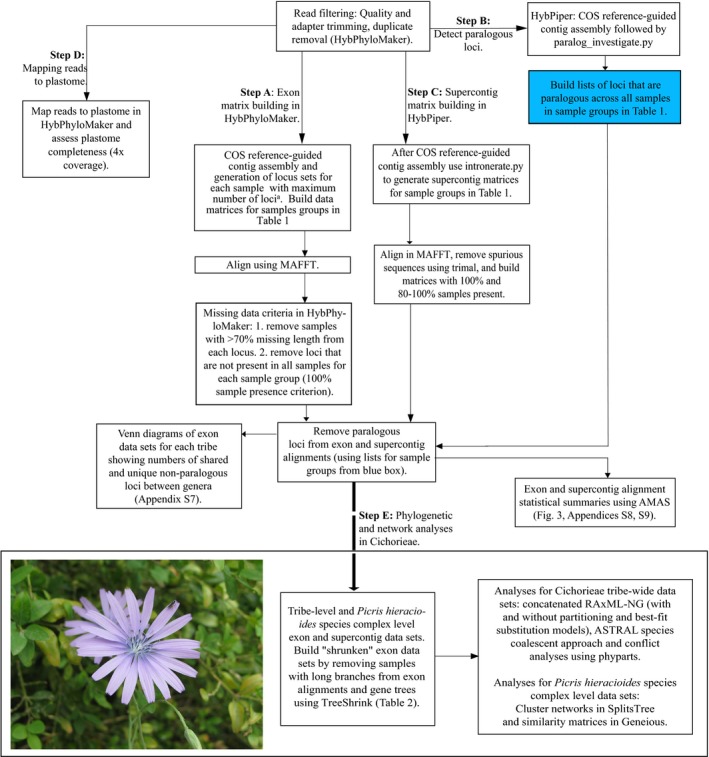
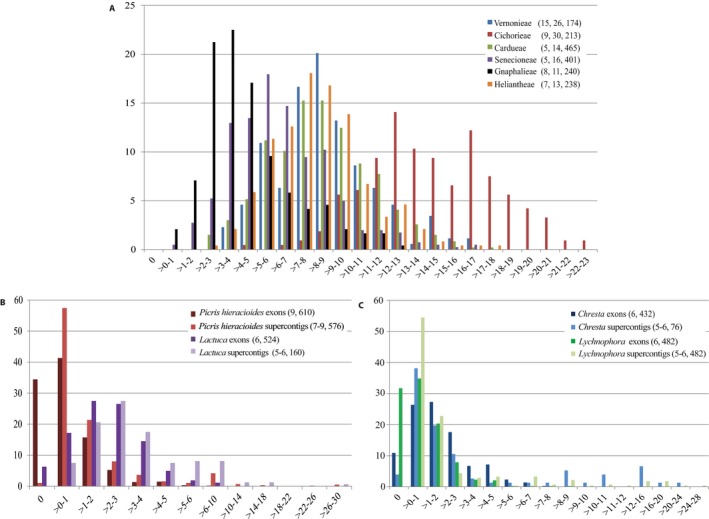
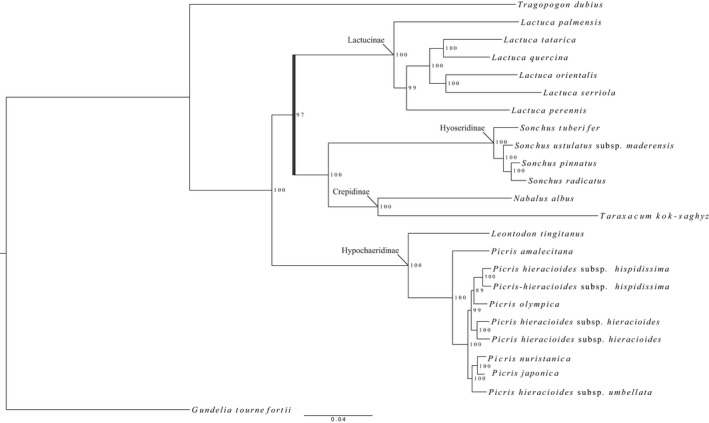
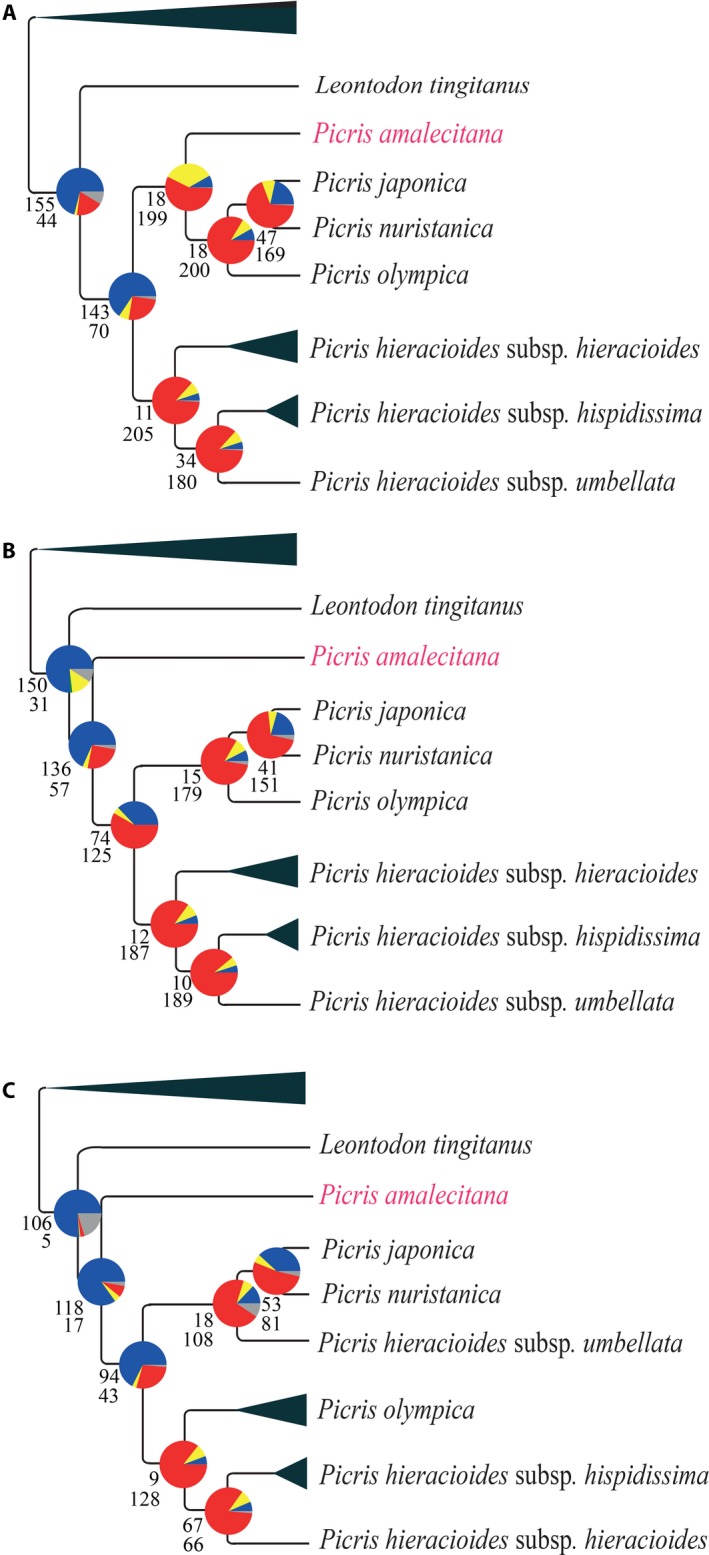
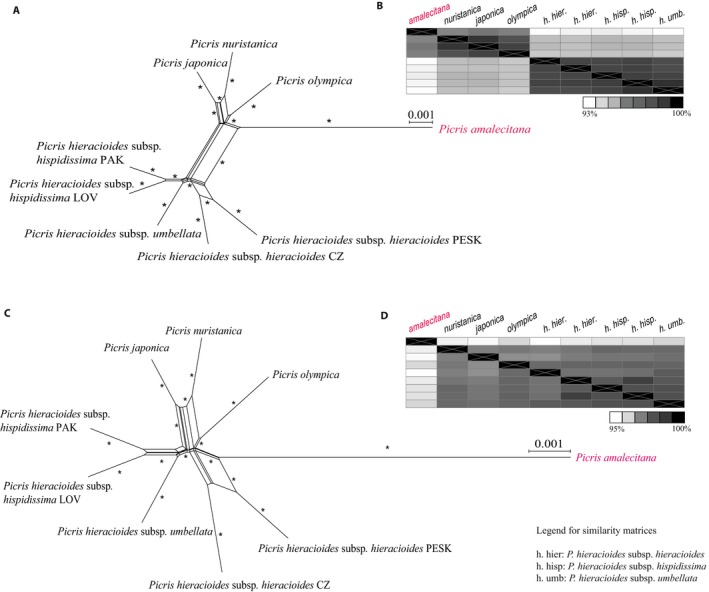
Hyb-Seq data from 112 Asteraceae samples were organized into groups at different taxonomic levels (tribe, genus, and species). For each group, data sets of non-paralogous loci were built and proportions of parsimony informative characters estimated. The impacts of analyzing alternative data sets, removing long branches, and type of analysis on tree resolution and inferred topologies were investigated in tribe Cichorieae.
Alignments of the Asteraceae family-wide Hyb-Seq locus set were parsimony informative at all taxonomic levels. Levels of resolution and topologies inferred at shallower nodes differed depending on the locus data set and the type of analysis, and were affected by the presence of long branches.
The approach used to build a Hyb-Seq locus data set influenced resolution and topologies inferred in phylogenetic analyses. Removal of long branches improved the reliability of topological inferences in maximum likelihood analyses. The Astereaceae Hyb-Seq probe set is applicable at multiple taxonomic depths, which demonstrates that probe sets do not necessarily need to be lineage-specific.
高通量测序杂交捕获技术(Hyb-Seq)是进化研究的有力工具。本文研究了菊科特异性Hyb-Seq探针集的适用性以及不同系统发育分析的结果。
将112个菊科样本的Hyb-Seq数据按不同分类水平(族、属和种)进行分组。对于每组,构建非旁系同源基因座数据集并估计简约信息特征的比例。在菊苣族中研究了分析替代数据集、去除长枝以及分析类型对树分辨率和推断拓扑结构的影响。
菊科全基因组Hyb-Seq基因座集的比对在所有分类水平上都具有简约信息。在较浅节点处推断的分辨率水平和拓扑结构因基因座数据集和分析类型而异,并受长枝的影响。
构建Hyb-Seq基因座数据集的方法影响了系统发育分析中推断的分辨率和拓扑结构。去除长枝提高了最大似然分析中拓扑推断的可靠性。菊科Hyb-Seq探针集适用于多个分类深度,这表明探针集不一定需要是谱系特异性的。