Wang Binran, Xiao Xiaoyue, Huang Fanwei, Liu Rong
College of Food Science and Technology, Nanjing Agricultural University, Nanjing 210095, China.
Department of Pathogen Biology, School of Basic Medicine, Huazhong University of Science and Technology, Wuhan 430030, China.
Antioxidants (Basel). 2019 Oct 30;8(11):522. doi: 10.3390/antiox8110522.
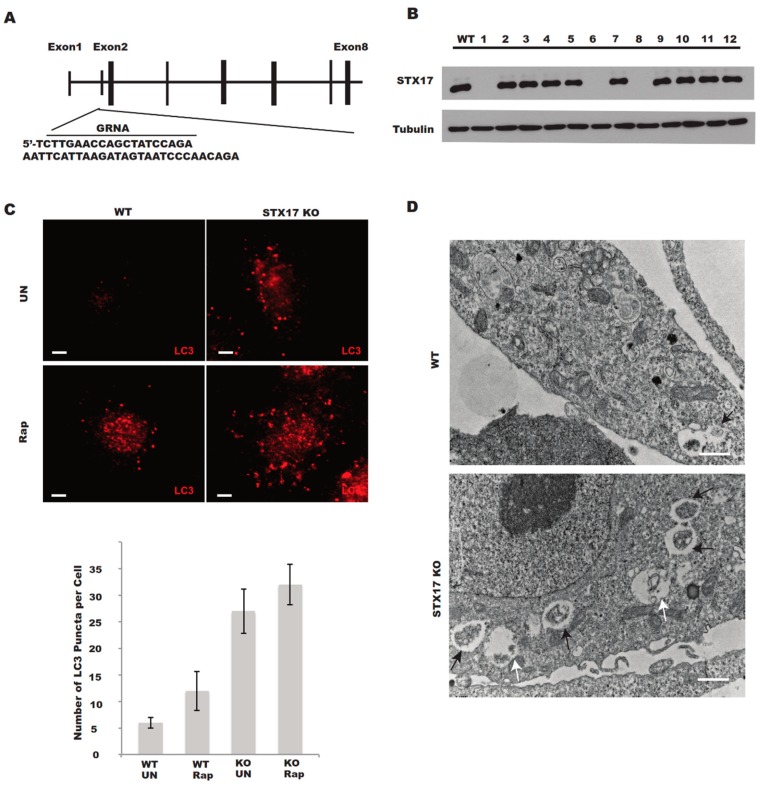
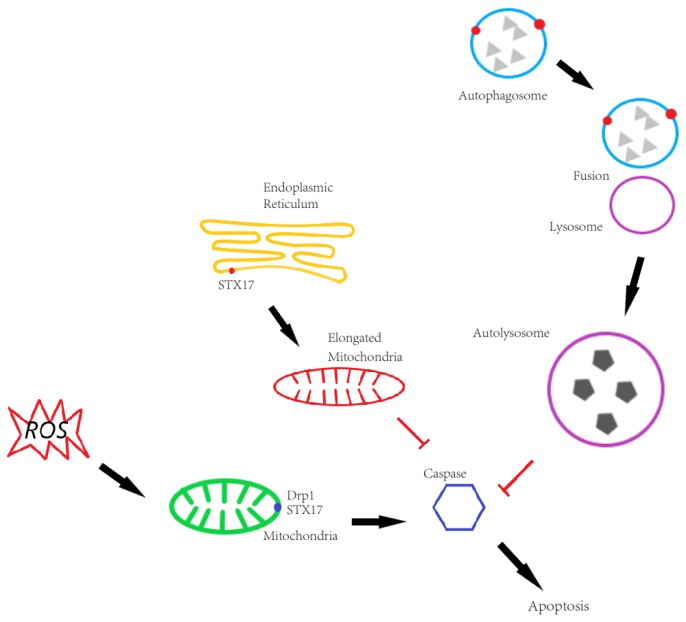
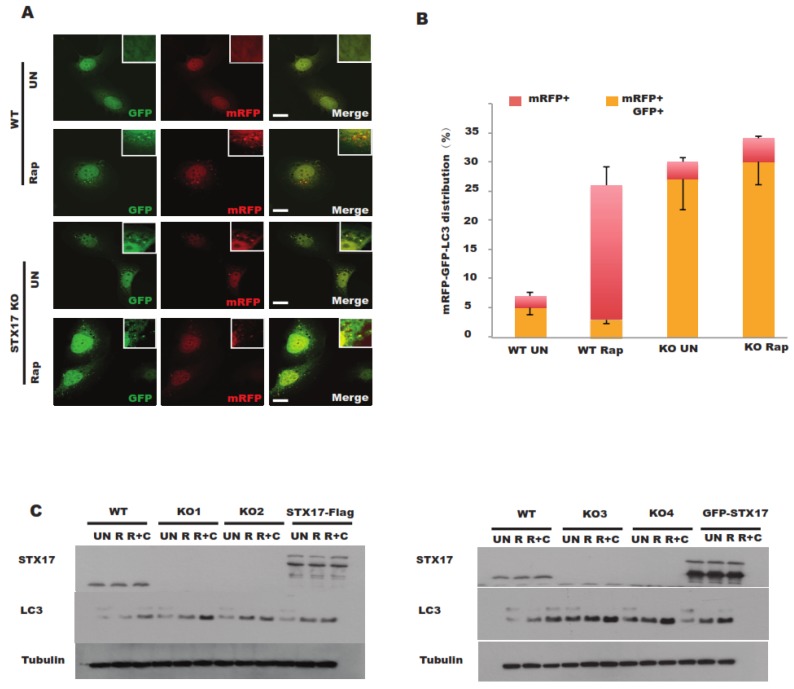
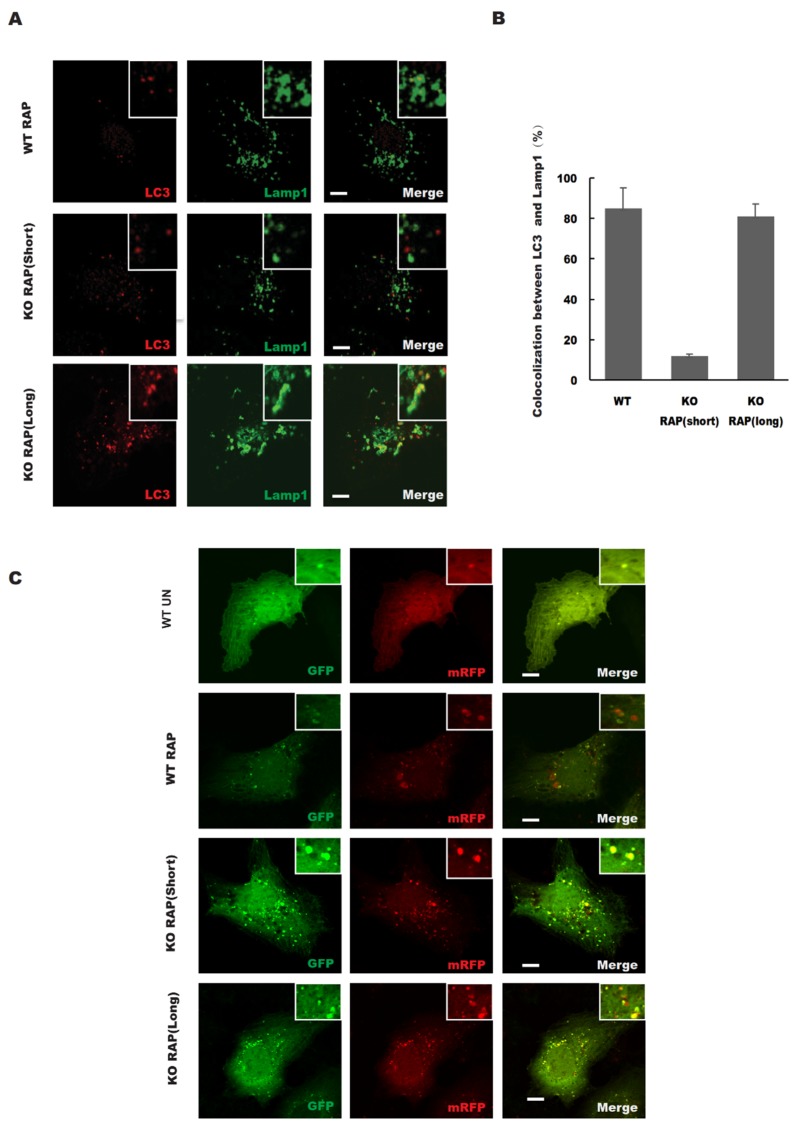
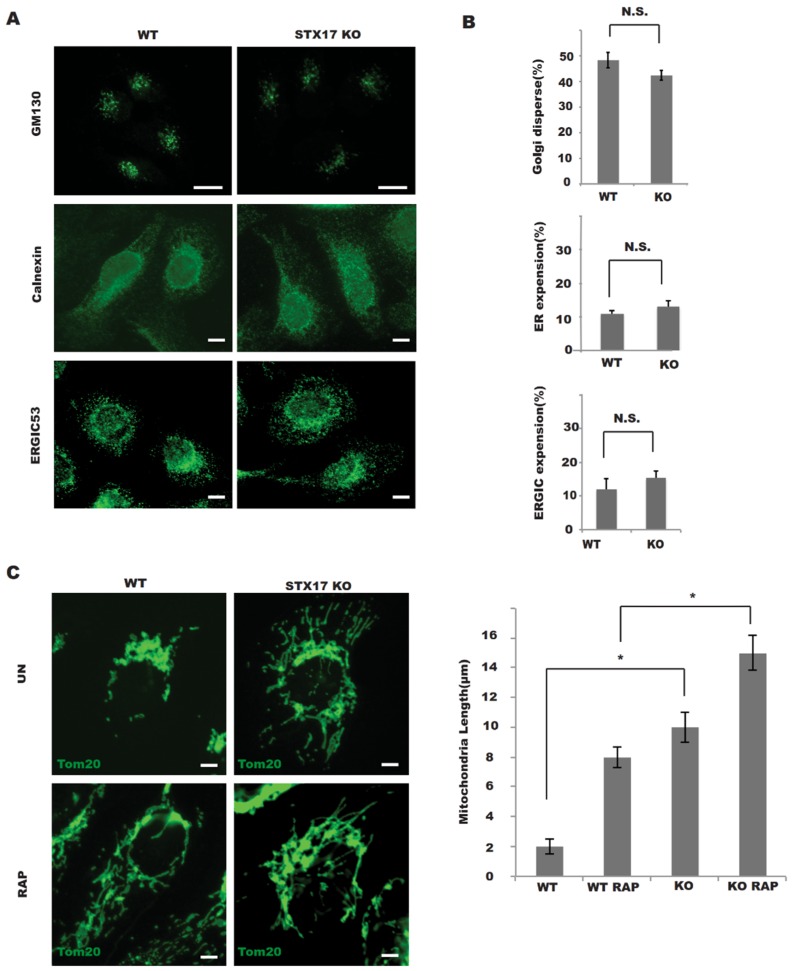
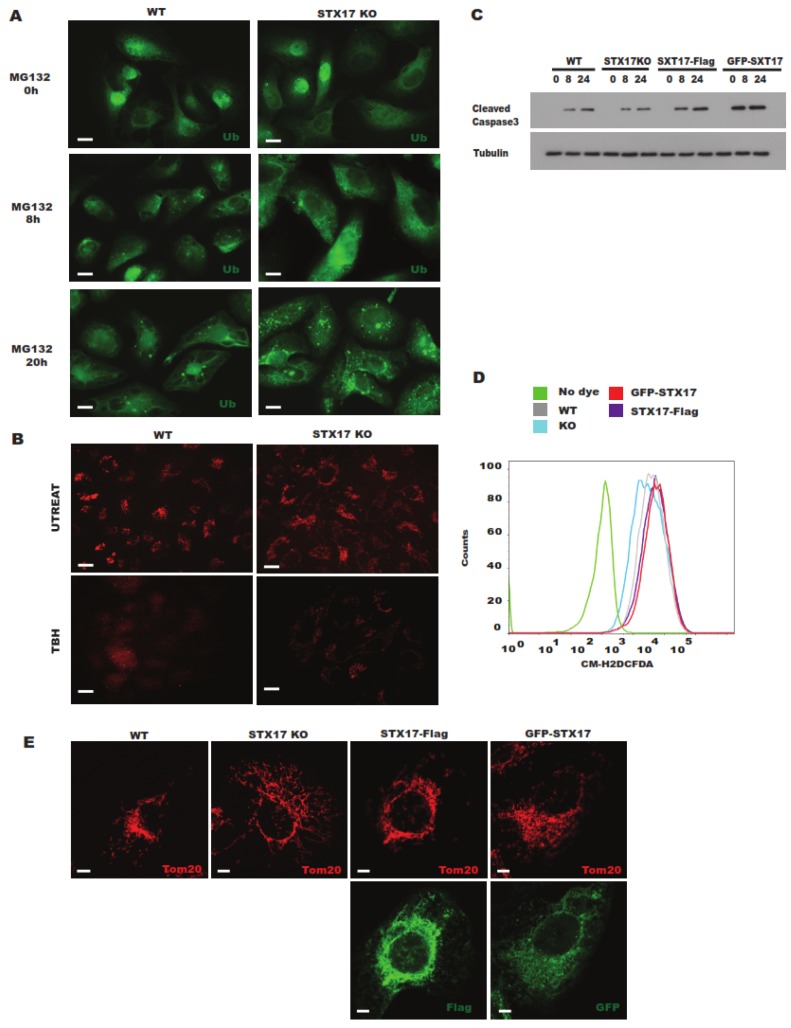
In this study, cell death induced by the oxidant tert-butylhydroperoxide (tBH) was observed in UOS cells; this phenotype was rescued by Syntaxin 17 (STX17) knockout (KO) but the mechanism is unknown. STX17 plays dual roles in autophagosome-lysosome fusion and mitochondrial fission. However, the contribution of the two functions of STX17 to apoptosis has not been extensively studied. Here, we sought to dissect the dual roles of STX17 in oxidative-stress-induced apoptosis by taking advantage of STX17 knockout cells and an autophagosome-lysosome fusion defective mutant of STX17. We generated STX17 knockout UOS cells using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system and the STX17 knockout cells were reconstituted with wild-type STX17 and its autophagosome-lysosome fusion defective mutant. Autophagy was assessed by autophagic flux assay, Monomer red fluorescent protein (mRFP)-GFP-LC3 assay and protease protection assay. Golgi, endoplasmic reticulum (ER)/ER-Golgi intermediate compartment (ERGIC) and mitochondrial dynamics were examined by staining the different indicator proteins. Apoptosis was evaluated by caspase cleavage assay. The general reactive oxygen species (ROS) were detected by flow cytometry. In STX17 complete knockout cells, sealed autophagosomes were efficiently formed but their fusion with lysosomes was less defective. The fusion defect was rescued by wild-type STX17 but not the autophagosome-lysosome fusion defective mutant. No obvious defects in Golgi, ERGIC or ER dynamics were observed. Mitochondria were significantly elongated, supporting a role of STX17 in mitochondria fission and the elongation caused by STX17 KO was reversed by the autophagosome-lysosome fusion defective mutant. The clearance of protein aggregation was compromised, correlating with the autophagy defect but not with mitochondrial dynamics. This study revealed a mixed role of STX17 in autophagy, mitochondrial dynamics and oxidative stress response. STX17 knockout cells were highly resistant to oxidative stress, largely due to the function of STX17 in mitochondrial fission rather than autophagy.
在本研究中,观察到氧化剂叔丁基过氧化氢(tBH)诱导UOS细胞发生细胞死亡;Syntaxin 17(STX17)基因敲除(KO)可挽救此表型,但其机制尚不清楚。STX17在自噬体-溶酶体融合和线粒体分裂中发挥双重作用。然而,STX17的这两种功能对细胞凋亡的贡献尚未得到广泛研究。在此,我们利用STX17基因敲除细胞和STX17的自噬体-溶酶体融合缺陷突变体,试图剖析STX17在氧化应激诱导的细胞凋亡中的双重作用。我们使用成簇规律间隔短回文重复序列(CRISPR)/CRISPR相关蛋白9(Cas9)系统构建了STX17基因敲除的UOS细胞,并用野生型STX17及其自噬体-溶酶体融合缺陷突变体重构了STX17基因敲除细胞。通过自噬通量分析、单体红色荧光蛋白(mRFP)-绿色荧光蛋白(GFP)-微管相关蛋白1轻链3(LC3)分析和蛋白酶保护分析评估自噬。通过对不同指示蛋白进行染色来检测高尔基体、内质网(ER)/内质网-高尔基体中间区(ERGIC)和线粒体动力学。通过半胱天冬酶切割分析评估细胞凋亡。通过流式细胞术检测一般活性氧(ROS)。在STX17完全敲除的细胞中,可有效形成封闭的自噬体,但它们与溶酶体的融合缺陷较少。野生型STX17可挽救融合缺陷,而自噬体-溶酶体融合缺陷突变体则不能。未观察到高尔基体、ERGIC或ER动力学有明显缺陷。线粒体显著延长,支持STX17在线粒体分裂中的作用,并且由STX17基因敲除引起的延长可被自噬体-溶酶体融合缺陷突变体逆转。蛋白质聚集的清除受损,这与自噬缺陷相关,但与线粒体动力学无关。本研究揭示了STX17在自噬、线粒体动力学和氧化应激反应中的混合作用。STX17基因敲除细胞对氧化应激具有高度抗性,这主要归因于STX17在线粒体分裂中的功能,而非自噬。