Monash Centre for Data Science, Faculty of Information Technology, Monash University, Melbourne, VIC, 3800, Australia.
Division of Cancer Epidemiology, Cancer Council Victoria, Melbourne, VIC, 3004, Australia.
BMC Bioinformatics. 2019 Nov 21;20(1):602. doi: 10.1186/s12859-019-3178-6.
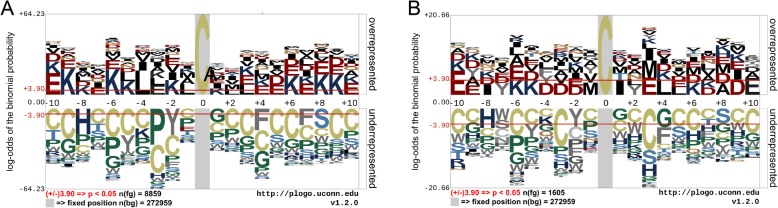
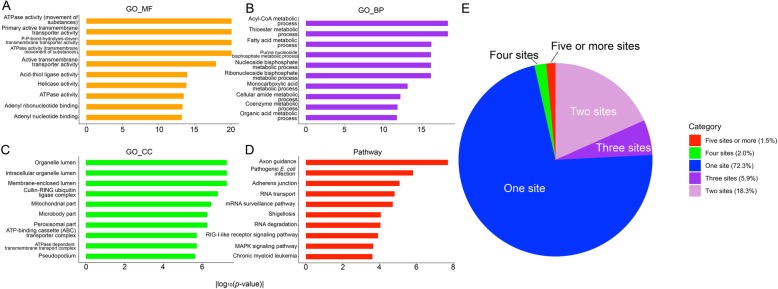
S-sulphenylation is a ubiquitous protein post-translational modification (PTM) where an S-hydroxyl (-SOH) bond is formed via the reversible oxidation on the Sulfhydryl group of cysteine (C). Recent experimental studies have revealed that S-sulphenylation plays critical roles in many biological functions, such as protein regulation and cell signaling. State-of-the-art bioinformatic advances have facilitated high-throughput in silico screening of protein S-sulphenylation sites, thereby significantly reducing the time and labour costs traditionally required for the experimental investigation of S-sulphenylation.
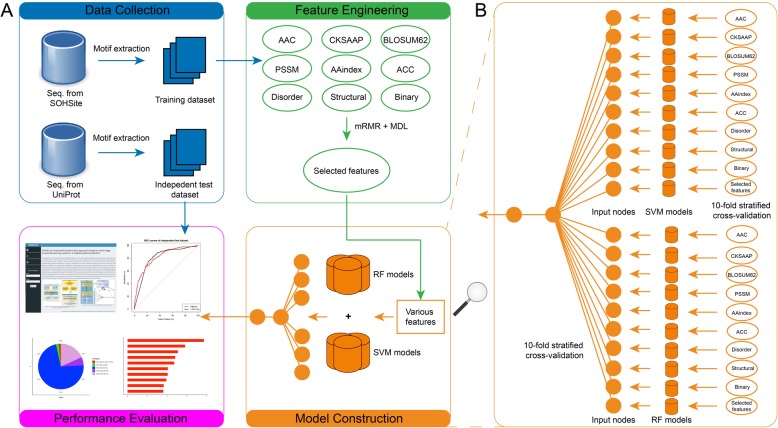
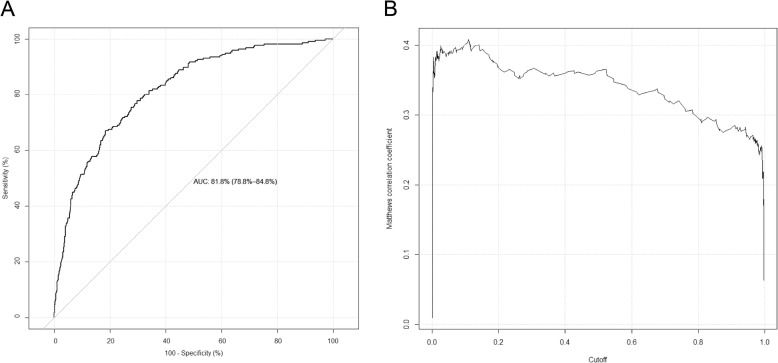
In this study, we have proposed a novel hybrid computational framework, termed SIMLIN, for accurate prediction of protein S-sulphenylation sites using a multi-stage neural-network based ensemble-learning model integrating both protein sequence derived and protein structural features. Benchmarking experiments against the current state-of-the-art predictors for S-sulphenylation demonstrated that SIMLIN delivered competitive prediction performance. The empirical studies on the independent testing dataset demonstrated that SIMLIN achieved 88.0% prediction accuracy and an AUC score of 0.82, which outperforms currently existing methods.
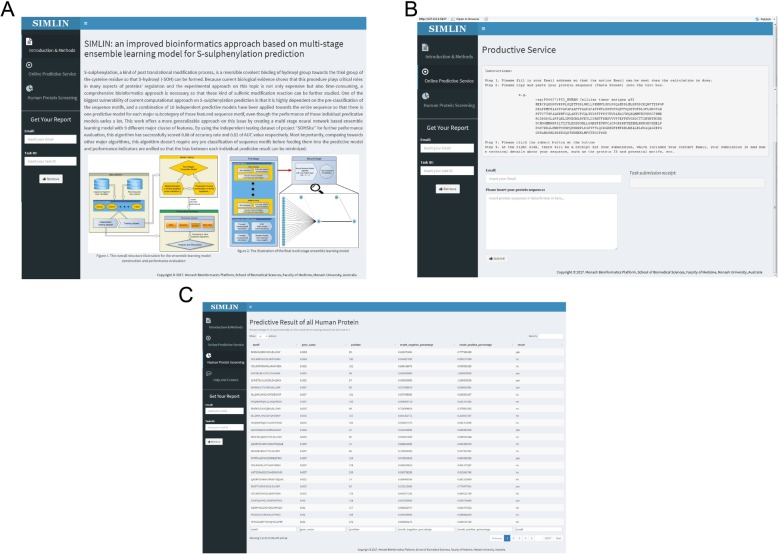
In summary, SIMLIN predicts human S-sulphenylation sites with high accuracy thereby facilitating biological hypothesis generation and experimental validation. The web server, datasets, and online instructions are freely available at http://simlin.erc.monash.edu/ for academic purposes.
S-亚磺酰化是一种普遍存在的蛋白质翻译后修饰(PTM),其中通过半胱氨酸(C)的巯基的可逆氧化形成 S-羟基(-SOH)键。最近的实验研究表明,S-亚磺酰化在许多生物功能中起着关键作用,如蛋白质调节和细胞信号转导。最先进的生物信息学进展促进了蛋白质 S-亚磺酰化位点的高通量计算机筛选,从而大大减少了传统上对 S-亚磺酰化进行实验研究所需的时间和劳动力成本。
在这项研究中,我们提出了一种新的混合计算框架,称为 SIMLIN,用于使用基于多阶段神经网络的集成学习模型准确预测蛋白质 S-亚磺酰化位点,该模型集成了蛋白质序列衍生和蛋白质结构特征。针对 S-亚磺酰化的最新预测器进行的基准实验表明,SIMLIN 提供了有竞争力的预测性能。在独立测试数据集上的实证研究表明,SIMLIN 实现了 88.0%的预测准确率和 0.82 的 AUC 得分,优于现有方法。
总之,SIMLIN 可以高精度地预测人类 S-亚磺酰化位点,从而促进生物学假设的生成和实验验证。该网络服务器、数据集和在线说明可在学术目的免费使用:http://simlin.erc.monash.edu/。