School of Computer Science and Technology, Harbin Institute of Technology, Harbin, 150001, China.
Department of Mathematics, Harbin Institute of Technology, Harbin, 150001, China.
BMC Bioinformatics. 2019 Nov 25;20(Suppl 18):573. doi: 10.1186/s12859-019-3132-7.
During procedures for conducting multiple sequence alignment, that is so essential to use the substitution score of pairwise alignment. To compute adaptive scores for alignment, researchers usually use Hidden Markov Model or probabilistic consistency methods such as partition function. Recent studies show that optimizing the parameters for hidden Markov model, as well as integrating hidden Markov model with partition function can raise the accuracy of alignment. The combination of partition function and optimized HMM, which could further improve the alignment's accuracy, however, was ignored by these researches.
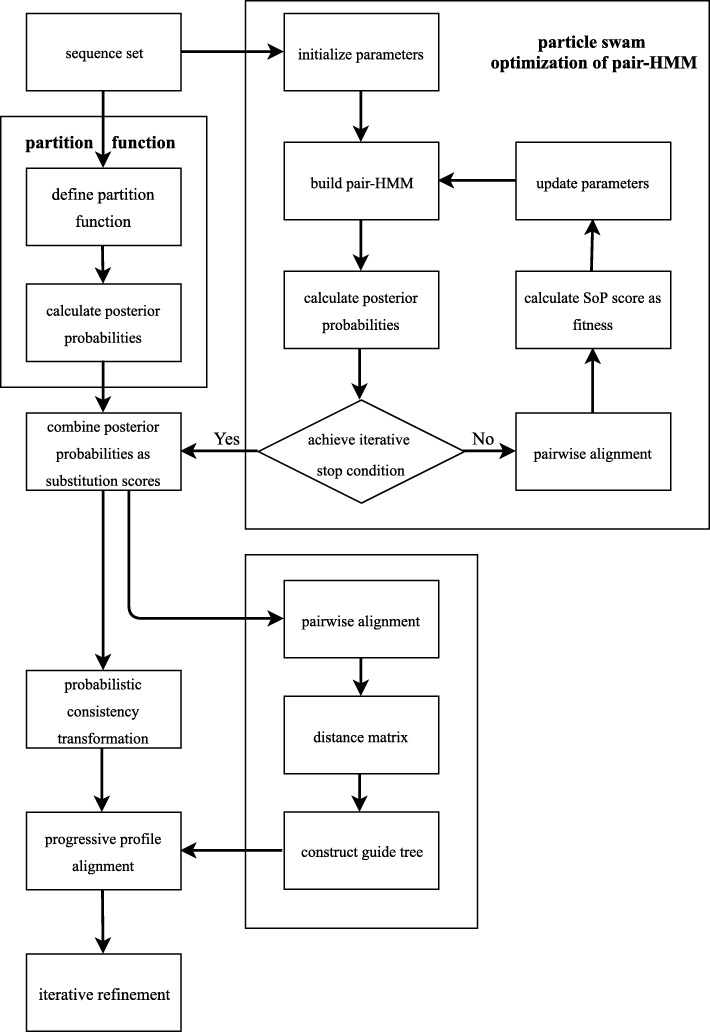
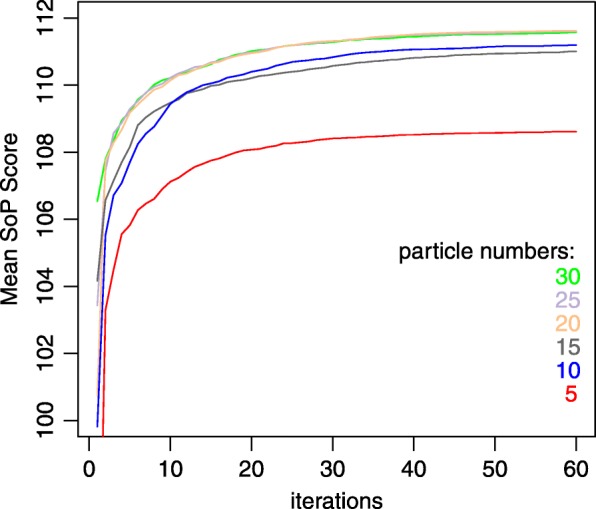
A novel algorithm for MSA called ProbPFP is presented in this paper. It intergrate optimized HMM by particle swarm with partition function. The algorithm of PSO was applied to optimize HMM's parameters. After that, the posterior probability obtained by the HMM was combined with the one obtained by partition function, and thus to calculate an integrated substitution score for alignment. In order to evaluate the effectiveness of ProbPFP, we compared it with 13 outstanding or classic MSA methods. The results demonstrate that the alignments obtained by ProbPFP got the maximum mean TC scores and mean SP scores on these two benchmark datasets: SABmark and OXBench, and it got the second highest mean TC scores and mean SP scores on the benchmark dataset BAliBASE. ProbPFP is also compared with 4 other outstanding methods, by reconstructing the phylogenetic trees for six protein families extracted from the database TreeFam, based on the alignments obtained by these 5 methods. The result indicates that the reference trees are closer to the phylogenetic trees reconstructed from the alignments obtained by ProbPFP than the other methods.
We propose a new multiple sequence alignment method combining optimized HMM and partition function in this paper. The performance validates this method could make a great improvement of the alignment's accuracy.
在进行多序列比对的过程中,使用序列比对的替换得分是非常重要的。为了计算自适应比对得分,研究人员通常使用隐马尔可夫模型或概率一致性方法,如分区函数。最近的研究表明,优化隐马尔可夫模型的参数,以及将隐马尔可夫模型与分区函数相结合,可以提高比对的准确性。然而,这些研究忽略了将分区函数与优化的隐马尔可夫模型相结合,以进一步提高比对的准确性。
本文提出了一种新的多序列比对算法 ProbPFP。它通过粒子群算法优化的隐马尔可夫模型与分区函数相结合。应用粒子群算法优化 HMM 的参数。然后,通过隐马尔可夫模型获得的后验概率与分区函数获得的后验概率相结合,从而计算出用于比对的综合替换得分。为了评估 ProbPFP 的有效性,我们将其与 13 种优秀或经典的 MSA 方法进行了比较。结果表明,在 SABmark 和 OXBench 这两个基准数据集上,ProbPFP 得到的比对结果具有最大的平均 TC 得分和平均 SP 得分,在 BAliBASE 基准数据集上,它的平均 TC 得分和平均 SP 得分排名第二。我们还将 ProbPFP 与其他 4 种优秀的方法进行了比较,通过基于这 5 种方法得到的比对结果,对从数据库 TreeFam 中提取的六个蛋白质家族的系统发育树进行重建。结果表明,参考树与基于 ProbPFP 得到的比对结果重建的系统发育树更为接近。
我们在本文中提出了一种新的多序列比对方法,将优化的隐马尔可夫模型与分区函数相结合。该方法的性能验证了其可以显著提高比对的准确性。