Department of Bioengineering, University of California, San Diego, La Jolla, CA, USA.
Department of Pediatrics, University of California, San Diego, La Jolla, CA, USA.
Nat Protoc. 2020 Jan;15(1):1-14. doi: 10.1038/s41596-019-0254-3. Epub 2019 Dec 20.
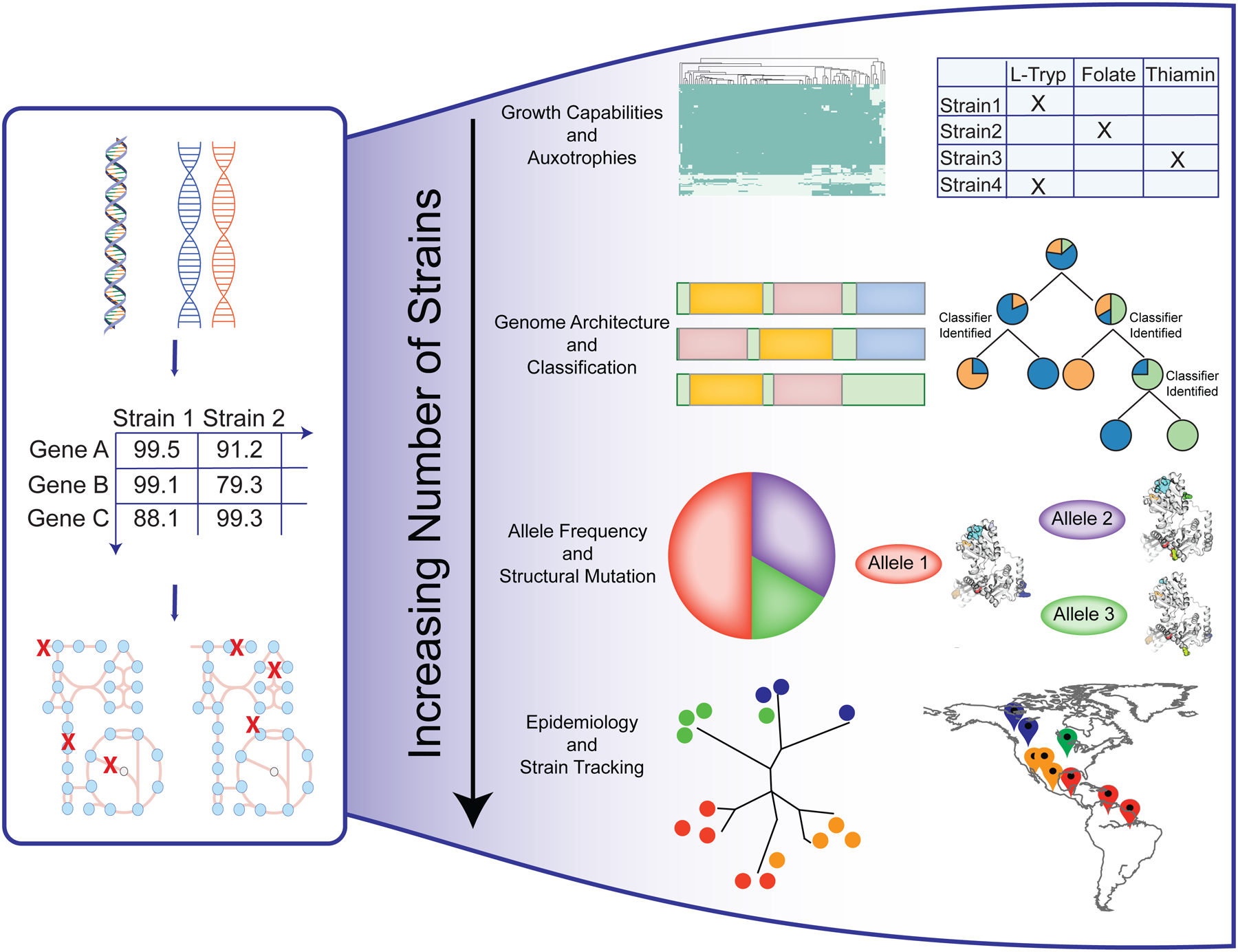
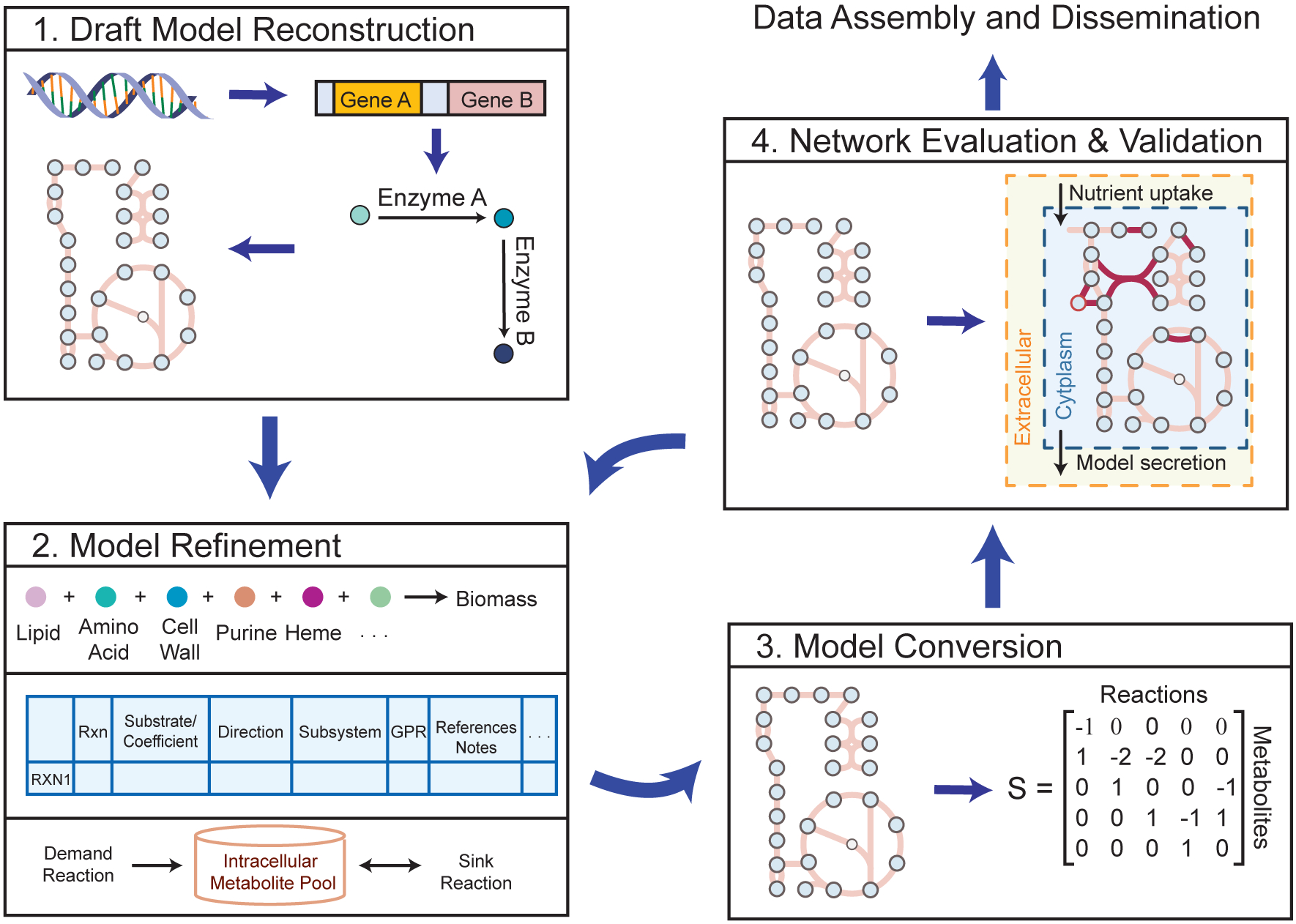
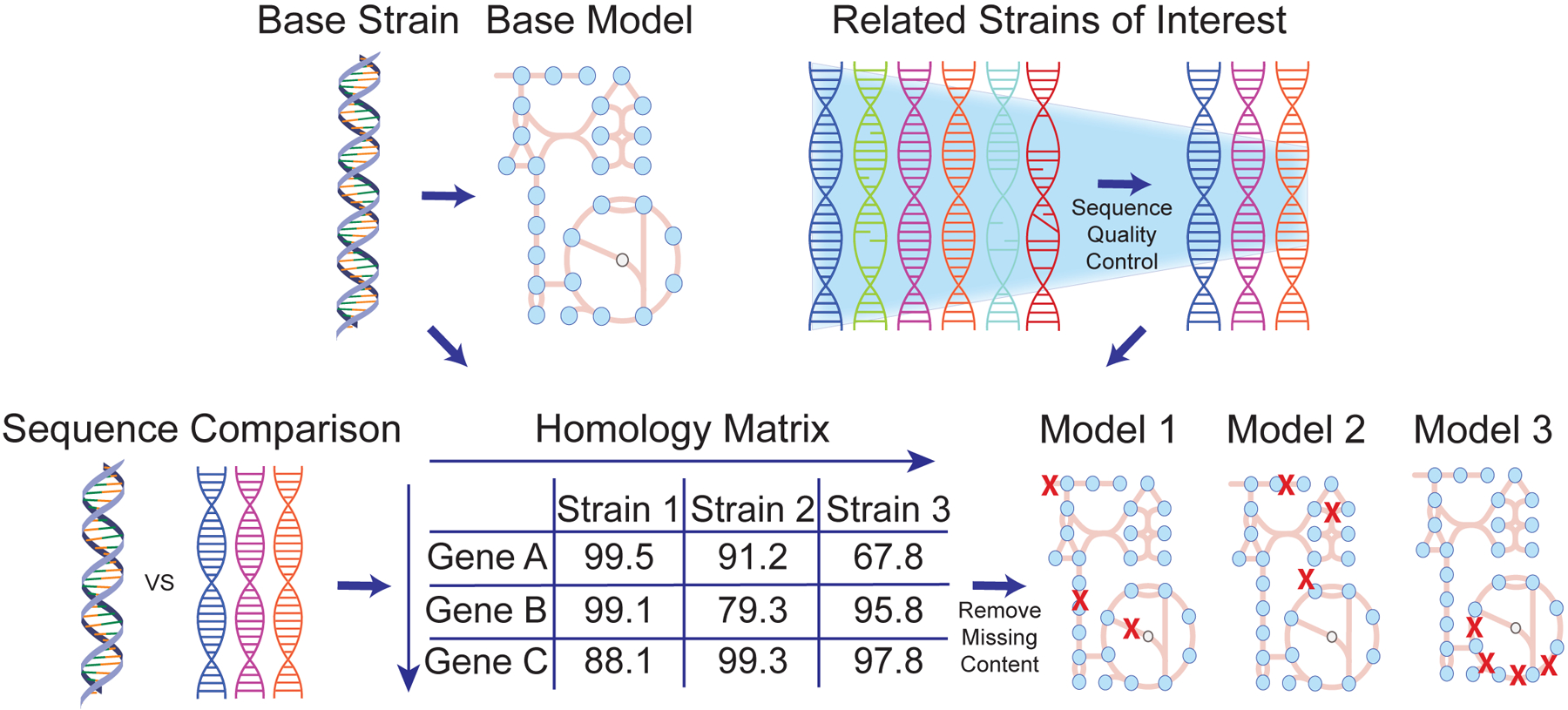
Genome-scale models (GEMs) of bacterial strains' metabolism have been formulated and used over the past 20 years. Recently, with the number of genome sequences exponentially increasing, multi-strain GEMs have proved valuable to define the properties of a species. Here, through four major stages, we extend the original Protocol used to generate a GEM for a single strain to enable multi-strain GEMs: (i) obtain or generate a high-quality model of a reference strain; (ii) compare the genome sequence between a reference strain and target strains to generate a homology matrix; (iii) generate draft strain-specific models from the homology matrix; and (iv) manually curate draft models. These multi-strain GEMs can be used to study pan-metabolic capabilities and strain-specific differences across a species, thus providing insights into its range of lifestyles. Unlike the original Protocol, this procedure is scalable and can be partly automated with the Supplementary Jupyter notebook Tutorial. This Protocol Extension joins the ranks of other comparable methods for generating models such as CarveMe and KBase. This extension of the original Protocol takes on the order of weeks to multiple months to complete depending on the availability of a suitable reference model.
在过去的 20 年里,人们已经构建并使用了细菌株代谢的基因组规模模型 (GEM)。最近,随着基因组序列数量呈指数级增长,多菌株 GEM 已被证明对定义物种特性很有价值。在这里,我们通过四个主要阶段,将用于生成单菌株 GEM 的原始方案扩展到能够生成多菌株 GEM:(i) 获取或生成参考菌株的高质量模型;(ii) 比较参考菌株和目标菌株之间的基因组序列,生成同源矩阵;(iii) 从同源矩阵生成菌株特异性模型草案;和 (iv) 手动编辑模型草案。这些多菌株 GEM 可用于研究跨物种的泛代谢能力和菌株特异性差异,从而深入了解其生活方式的范围。与原始方案不同,该过程具有可扩展性,并且可以使用补充的 Jupyter 笔记本教程部分自动化。该方案扩展与其他用于生成模型的可比方法(如 CarveMe 和 KBase)一起加入。该原始方案的扩展需要数周到数月的时间才能完成,具体取决于是否有合适的参考模型。